

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ORIGINAL ANTIGENIC SIN
In an effort to develop effective vaccines against pathogens that severely affect humans, the concept of original antigenic sin (OAS) has become a guiding light, making sense of baffling observations on the infection profiles of foreign pathogens and vaccine efficacies in humans. OAS was first described by Davenport et al. to explain antibody responses that were generated after influenza virus infection in people of different age groups (1). It was then suggested that antibody-forming mechanisms appear to be directed by initial childhood infections, such that exposure to antigenically related strains later in life results in progressive reinforcement of the primary antibody. According to this suggestion, OAS might 'freeze' the immune repertoire against a pathogen (2).
The repertoire freeze mechanism of OAS is understood as part of the hard-coded immune memory responses against a potential recurring pathogen (3). Recent research has shown that though it appears to be pathogen-dependent, such a memory response is also found during innate immune responses, and is not limited to adaptive immunity (4). This mechanism would protect the host from the pathogen like a well-trained defense team. Immunity against measles, mumps, and smallpox, among others, is a good example of memory responses that provide lifetime protection after a single natural infection or an effective vaccination (45). Unfortunately, most pathogens adapt to this immune program by constantly changing their surface molecules. The reason why OAS is stigmatized as 'sin' is its interference with the naïve immune response against variants of the pathogen (6). In this review, we introduce the concept of OAS as a type of 'immune repertoire freezing', the result of which can have both positive and negative impacts on immune responses against pathogens and on the course of diseases caused by such agents.
Go to : 
MECHANISM OF ORIGINAL ANTIGENIC SIN
The epitome of memory responses in the immune system is the rapid production of antibodies of different isotypes and effector cytokines such as IFN-g or interleukins, by antigen specific B lymphocytes (B cells) and the cognate thymus-derived lymphocytes (T cells), respectively (7). Phenotypically defined memory B and T cells have been shown to divide more rapidly than naive cells (789).
OAS, a process of repertoire freeze in memory responses (3101112) are shown in Fig. 1. When an antigen specific receptor (BCR) on the outer surface of B cells recognizes its cognate antigen of a pathogen, the B cell captures and processes the pathogen by presenting pieces of the antigen onto major histocompatibility complex class II (MHC II) molecules. A single B cell recognizes a single antigen on a pathogen, but can present as many as antigens of the pathogen on MHC II molecules for recognition of CD4+ T cells via a cognate T cell receptors (TCRs). Through this interaction, both antigen-specific B and T cells can proliferate and differentiate into plasma or memory B cells and effector or memory T cells, respectively, depending on environments of the interaction (131415). When an antigenic variant of the same pathogen appears, it is likely to be captured by memory B cells, which can recognize a conserved or cross-reactive recognition site (epitope) of the pathogen rather than by a naïve B cells. This is because memory B cells are expanded clones against the same specific antigen from the previous exposures to the antigen, whereas naïve B cells originate from unique VDJ recombinations, resulting in a repertoire that might not efficiently respond to a specific epitope. Before affinity maturation through a somatic hypermutation process, in part mediated by T cells, the affinity of the target to the BCR on the naïve B cells is generally low (16). Therefore, even though antigen-specific naïve B cells recognize a dominant antigenic site, they might not have an advantage in terms of competition for antigen capture over cross-reactive memory B cells. Furthermore, memory immune responses might help to rapidly clear the pathogen through limiting the availability of the antigen for variant specific naïve B cells (1718). Serum antibody, Fc receptor (FcR)-mediated non-specific capture, or presentation of the MHC II antigens by antigen presenting cells (APCs) bearing other FcR, such as macrophages and dendritic cells (DCs) (19), are also likely to further enrich antigen-specific memory T cells from a previous exposure, rather than antigen-specific naïve T cell activation, This occurs for the previously stated reason, specifically, that of better availability of memory T cells than antigen-specific naïve T cells.
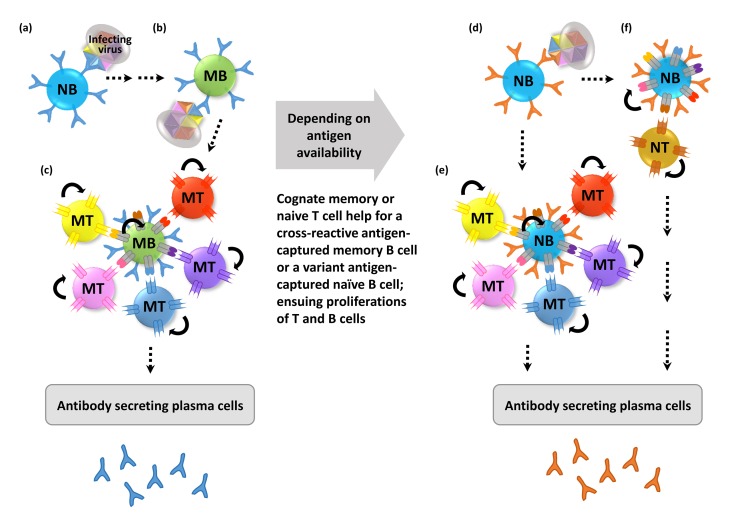 | Figure 1Conceptual mechanism of antigenic original sin. Surface antigens of a pathogen are designated by different colors. A naïve B (NB) cell specific for 'blue' antigen (with 'blue' receptors) recognizes a surface antigen on a pathogen designated by 'blue' (a). Potential antigen relay from DCs, and cognate T cells help on the way to memory B (MB) cell is omitted. A surface variant pathogen may be captured more readily by an abundant cross-reactive 'blue' specific memory B cell (b) than a naïve B cell. Subsequently, the cross-reactive 'blue' memory B cell presents the MHC II epitopes of the pathogen to the cognate memory T (MT) cells (c). Concomitant proliferation of the engaged memory B and T cells further reinforces the existing memory leading to repertoire freeze due to AOS. When the pathogen persists, a variant 'brown' antigen-specific naïve B cell might have an opportunity to capture the pathogen (d), and present the MHC II epitopes of the pathogen most likely to the cognate memory T (MT) cells (e), due to other conserved epitopes in the pathogen, rather than the cognate naïve T cells (f). All possible interactions depicted in (c) and (e) may not necessarily occur at the same time. Only one T cell engagement is depicted in (f) for simplicity, but it can be like (c) and (e).
|
As shown in Fig. 1, it is conceivable that a dominant antigenic site-specific naïve B cells happen to encounter the cognate epitope before the clearance of the pathogen by the cross-reactive memory B cell responses. This would be followed by activation of cognate memory T cells from previous exposures. Although the dominant response is repertoire freeze, antibody diversification may occur against different influenza strains through time (1), which can suggest that the frozen repertoire can melt and be imbued with some diversity. With regard to the memory immune response, increased diversity is an important concept for vaccine design. While OAS is harnessed by many universal influenza vaccine strategies (2021222324), OAS-mediated generation of cross-reactive, non-neutralizing antibodies against pathogens such as dengue virus can be a cause of enhanced disease severity after the second infection, which represents a hurdle for vaccine development (22526).
Go to :
ORIGINAL ANTIGENIC SIN AND VACCINE EFFICACY
The conceptual aim of a universal vaccine is to achieve optimal memory B cell and/or T cell responses toward a conserved epitope of a recurring and constantly changing pathogen, through a single immunization. However, it appears natural for the pathogen to change its surface antigenic region for survival, because the region typically harbors an immunodominant epitope, which poses a dilemma for universal vaccine strategies. In Table I, we summarize, albeit not extensively, the aspects of viral vaccine design as they relate to mechanisms of OAS, assuming recurring infections by the same pathogen with antigenic variation.

Table I
Conceptual effects of OAS on the immune responses generated through different vaccine preparation methods
![]()
Overcoming OAS is an important issue in terms of a vaccine development against recurring pathogens such as seasonal influenza viruses, because a vaccine strategy stimulating repertoire diversification would change the efficacy of the vaccine upon the appearance of variant strains. Conceivably, a repertoire freeze can be somewhat overcome by removing the limitation of antigen availability, even though there can be practical limitations to this. The use of more antigens for an inactivated influenza vaccine was shown to be associated with higher influenza-specific antibody titers in the serum of vaccinated HIV patients, compared to that observed with a lower number of antigens (27). A cohort study of healthy adults vaccinated with a virus-like, virosomal, trivalent inactivated influenza vaccine (TIV; including antigens from H1N1, H3N2, and influenza B viruses) or with a split TIV showed higher influenza-specific IgM responses with the split vaccine cohort compared to that with the virosomal vaccine cohort (28). In the same study, HIV patients in the virosomal vaccine cohort showed qualitatively similar immune responses as healthy subjects, and a difference was only quantitatively identified. IgM is the first antibody isotype secreted by short-lived plasma cells, and is generated in the initial proliferation and expansion phase in antigen-specific naive B cells, before a T cell-mediated isotype class switch occurs (29). Because of this, IgM responses might be considered representative of naïve immune responses. Similarly, there have been reports demonstrating that a live-attenuated influenza vaccine (LAIV) is more effective in children, whereas the split TIV is more effective in adults (303132). It is likely that the split TIV could allow the immune system of adults, harboring the accumulated effects of OAS, to expand its diversity, compared to that with LAIV. Split TIV can minimize the OAS-dominated 'winner takes all' situation in the competition for antigen capturing (Fig. 2). In the case of split/subunit vaccines for adults, some subdominant but conserved epitopes can be exposed to naïve or memory B cells that can be activated by cognate memory T cells. Although no DNA vaccine has been licensed against influenza, it could potentially work like split/subunit and live vaccines. If such DNA is for non-specific expression and for the release of encoded viral proteins, its vaccine effects will theoretically be similar to those of split/subunit vaccines. For DNA delivery, targeting to APCs, memory B cells, and T cells can be achieved, which could result in the LAIV-like effects without the aforementioned 'winner take all' competition for antigens, which might eventually support repertoire diversification. It appears that the vaccine preparation method can determine the magnitude of naïve immune responses, or in other words, repertoire diversification, as shown in Table I. Such repertoire diversification efforts might give a chance to the dominant epitopes of a predicted variant, which could undergo further variation. However, in the context of immune memory generated by such strategies against conserved subdominant epitopes, whether corresponding conserved epitope-specific memory B cells will be able to gain access to the antigen beyond dominant epitope cross-reactive memory B cells in the event of a new variant infection (3334), is another issue (Fig. 3).
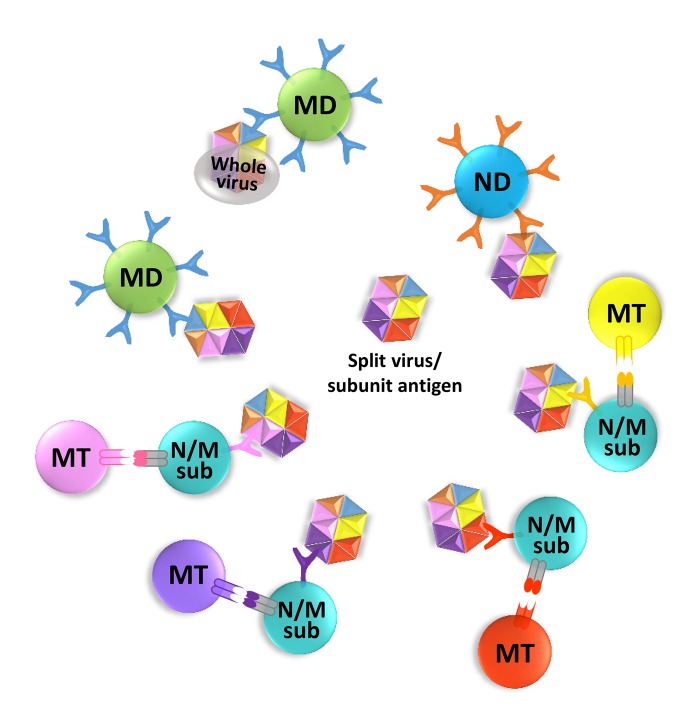 | Figure 2Antigen accessibility in different methods of vaccine preparation. Conceptual depiction of different epitope accessibilities on a whole virus and on a soluble protein. A surface variant whole virus may be captured most readily by the cross-reactive memory (MD) B cells recognizing the well exposed dominant epitope, thus reinforcing the memory of the dominant cross-reactivity by AOS. Split inactivated virus or subunits may expose conserved subdominant epitopes, normally not well exposed in a whole virus, to each subdominant epitope specific naïve or memory (N/Msub) B cell. Dominant or subdominant epitope specificity is symbolically shown with more or less number of receptors. The receptor of an antigen specific B cell and the cognate antigen are depicted in the same color. Only a single interacting cognate memory T (MT) cell to each B cell is depicted for simplicity. Dominant epitope recognizing B cell responses are the same as in Fig. 1.
|
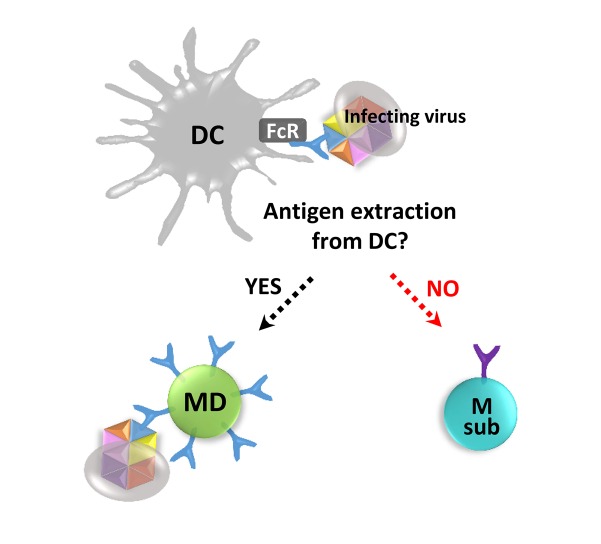 | Figure 3Antigen capture in the recall response. Antigen capture in the recall response is conceptually depicted. AOS-reinforced cross-reactive serum antibodies against a dominant epitope of infecting surface variant virus may participate in FcR-mediated antigen capture by APCs. In the recall response, the cross-reactive memory B cell recognizing the exposed dominant epitope (MD) can capture the whole viral antigen directly or via a dendritic cell (DC), and its cross-reactive memory becomes reinforced. However, a memory B cell recognizing a poorly exposed conserved subdominant epitope (Msub) will have difficulty in binding the whole virus antigen, and the conserved subdominant B cell memory may be suppressed.
|
Go to :
ORIGINAL ANTIGENIC SIN AND INFLUENZA VIRUS IMMUNITY: 'GOOD' OAS?
Regardless of the methods of vaccination, upon exposure to an unpredicted variant pathogen, clearance may occur through a 'good' OAS or, a 'good' OAS and/or the development of a diversified immune repertoire from naïve immune responses against the variant epitopes by prolonged exposure to the pathogen. This implies that the pathogen could have enough time to grow and harm the host before being cleared.
The evidence for 'good' (or beneficial) OAS is difficult to discern. In the cases of influenza vaccination, a mismatched vaccine is not 'optimally' protective, although the mismatch often stems from a difference of only a few amino acids in the globular heads of the hemagglutinin between the vaccine strain and the circulating strain (353637). However, as observed during the 2009 H1N1 influenza pandemic, even if mismatched, vaccination with a TIV (including seasonal H1N1 virus components) in the previous season proved to be beneficial, as unvaccinated individuals suffered more severe disease outcomes (31). This suggests that marginal but beneficial cross-reactivity from OAS can help to fight against the influenza virus.
The 2009 H1N1 influenza pandemic helped us gain a better understanding of immune responses against influenza virus infection. When donor sera taken before and after seasonal influenza vaccination (2007-08 or 2008-09 season) were analyzed for cross-reactivity to the pandemic 2009 H1N1 influenza virus (p2009 H1N1), adults older than 60 were shown to have a high rate of preexisting immunity (38). Interestingly, the sera of older adults taken before seasonal vaccination showed a higher microneutralization antibody titer against p2009 H1N1 than against the seasonal H1N1; in addition, the titer against p2009 H1N1 increased slightly after seasonal vaccination, which was understandably not to the level of antibody titer increase against seasonal H1N1. Individuals born closer to 1918 showed a higher protective microneutralization titer against p2009 H1N1 (38), suggesting that specific humoral immunity against the influenza virus can be very long lasting. It is very likely that immune memory to the conserved epitope(s) of the 1918 and p2009 H1N1 strains has been continuously boosted through years of infections with variant H1N1 strains through the mechanism of OAS. Although impossible to confirm, and as suggested by the need for seasonal vaccinations, it is very likely that the continually boosted 1918 H1N1 OAS memory is beneficial but not sufficient to defend against continuously changing seasonal variants. Such immune memory was fully protective (assuming that microneutralization is indicative of protection against the virus in vivo) only when similar viruses were presented.
So far, we have discussed cases suggesting the beneficial effects of OAS against influenza viruses. However, it is apparent that continuous boosting of conserved epitopes through recurrent infections by variant viruses, does not offer complete protection against a new variant infection; although it sounds like a circular argument, because of lack of protection, infections recur. Otherwise, seasonal vaccination against new variants of the influenza virus would be unnecessary (39).
The challenge of a universal vaccine design appears to be the optimization of the 'good' OAS. A memory B cell repertoire specific for difficult-to-access subdominant conserved epitopes of influenza virus cannot be adequately generated in the first place, or not recalled efficiently, or potentially have negative effects on the naïve immune responses against a particular variant. We discussed previously that the generation of a memory B cell repertoire against conserved but difficult-to-access epitopes could be conceptually achieved through a suitable vaccine preparation method. However, recall efficiency may be another issue (Fig. 3); the potential for this repertoire to contribute to adverse outcomes during influenza virus infection has not been adequately studied. Universal vaccine approaches have yielded promising results against influenza in animal models (20222324). However, animal models cannot appropriately and exhaustively model the history of vaccination and infection in humans.
Go to :
CAN 'BAD' OAS PLAY A ROLE IN INFLUENZA VIRUS INFECTION?
Comparison of the two H1N1 influenza pandemics of 1918 and 2009 appears to be educational. In terms of diseases severity, the two pandemics are incomparable; while some 50 million deaths were estimated in the 1918 pandemic, the estimated number of influenza-related deaths during the 2009 pandemic was no higher than yearly seasonal influenza-related deaths (40). In the case of the 2009 pandemic, although in vitro assays showed marginal cross-reactivity between the sera of seasonal TIV-vaccinated healthy adults and p2009 H1N1, seasonal vaccination was associated with less severe disease outcomes (31). This suggests that some protection was afforded either by the non-neutralizing antibodies against the conserved epitopes potentially through the FcR-mediated protective responses (41), or by the memory T cell responses against the conserved epitopes, or by both. In the case of 1918, records revealed that more young adults (presumably with robust health) succumbed to the virus than the children and the elderly individuals (42), suggesting that OAS might have resulted in adverse effects in immunocompetent adults and that potentially beneficial immune memory in the elderly was protective. Interestingly, in young adults, p2009 H1N1 resulted in a 2~4-fold greater risk of severe outcomes, specifically those requiring intensive care and mechanical ventilation, compared to seasonal influenza (43). This suggests that the 1918 pandemic was actually recapitulated on a small scale during the 2009 pandemic.
Obviously, the 1918 pandemic was not the first human encounter with the influenza virus (44). We can assume that individuals must have had some level of immunity against the influenza virus through recurring natural infections, which could be considered a form of whole virus vaccination. From such type of vaccination, we can conceptually assume (Fig. 1 and Table I) that people in 1918 had very little B cell memory against conserved but subdominant epitopes of the influenza virus, but undoubtedly had some CD4 and CD8 T cell memory against conserved epitopes of the virus. If OAS had mediated an adverse immune response, involved in the severity of the disease caused by the 1918 H1N1 virus infection, apart from the high virulence of the virus (45), the key contributing factors must have been cross-reactive non-neutralizing memory B cell responses against a dominant epitope or memory T cell responses against conserved epitopes.
An important question is whether memory CD4 and CD8 cells that recognize conserved epitopes cause harmful effects in response to an infective viral variant. Protection against the influenza virus through CD4 and CD8 T cell immunity alone was demonstrated in mice (4647). However, although memory CD4 and CD8 cells persist for a long time through homeostatic proliferation (1048), cross-protective resident memory CD8 T cells in pulmonary tissues, which are specific for the influenza virus, were shown to be short-lived (495051). These observations from experiments using mice have limitations; however, they appear to partly explain why infections recur, regardless of conserved epitopes of internal influenza proteins that can potentially generate universally protective memory CD8 T cells. Memory CD4 T cells against conserved epitopes can conceptually function as a "double-edged sword" when faced with a variant; the cross-reactive OAS responses of memory B cells against dominant epitopes, with the help of memory CD4 T cells recognizing conserved epitopes, can be beneficial or damaging (i.e. 'good' OAS or 'bad' OAS).
Whether a non-neutralizing cross-reactive OAS response, with the help of memory CD4 T cells against conserved epitopes, contributed to such devastating manifestations of the1918 H1N1 pandemic H1N1 (5253) is a matter of speculation. However, the well-documented effect of 'bad' OAS in dengue virus infection could provide some clues for this issue.
Go to :
ORIGINAL ANTIGENIC SIN AND DENGUE VIRUS INFECTION: CLASSIC 'BAD' OAS
Infection with a dengue virus can be asymptomatic or result in dengue fever (DF) or dengue hemorrhagic fever (DHF). Substantial evidence has associated DHF with secondary infection of a serotype of the dengue virus different from the virus to which an individual has already been exposed. The most widely accepted hypothesis for the pathogenesis of DHF is antibody-dependent enhancement (ADE) of dengue virus infection (reviewed in (25)).
ADE depends on pathogen binding antibodies and antibody binding complement proteins and their receptors such as FcRs and complement receptors. Therefore ADE is observed mainly with pathogens that efficiently infect FcR-bearing myeloid lineage cells, such as dendritic cells and macrophages (2). DC-SIGN is a universal receptor for the dengue virus, and DC-SIGN-expressing immature DCs were shown to be the initial site of infection (54). Dengue virus can also infect other FcR-bearing cells of the immune system, such as mature DCs and macrophages. Dengue virus-infected human endothelial cells are capable of antibody-dependent complement activation and undergo apoptosis (55). Although conceptually feasible, it is not clear whether the dengue virus-antibody-complement-complement receptor complex on the cell surface can actually enhance infection of dengue virus in endothelial cells. However, a similar ADE mechanism has been observed during infections with other viruses like HIV (2). It has been shown that human endothelial cells are highly susceptible to dengue virus infection (56). Uninfected endothelial cells might also participate in the disease process via complement receptors on endothelial cells and platelets that can mediate inflammatory responses (5758); however, the infected endothelium, in the presence of cross-reactive antibodies, might resemble the endothelium of a host experiencing graft rejection (59). ADE by itself is not thought to correlate with disease severity and DHF; complement activation by virus-antibody or antigen-antibody complexes is a feature of severe dengue infection, and it is temporally related to plasma leakage (6).
There are four serotypes of dengue viruses. Immune responses against dengue viruses are not very different from immune responses against other viruses such as influenza virus. There are conserved features and differences among the variants, and accordingly, inevitable OAS can occur. An individual infected with one type of dengue virus never succumbs to the same type of dengue virus infection because of its long-lasting homotypic protective immunity against the primary strain (6). However, when the same individual is infected with different serotype of dengue virus, serotype cross-reactive memory B and T cells from the prior exposure become activated. However, these cross-reactive responses are not protective, and are even detrimental. It was shown that highly cross-reactive monoclonal antibodies against dengue viruses could be potently neutralizing at an optimal concentration but resulted in ADE at a suboptimal concentration (60). It appears that cross-reactive antibodies are not necessarily inherently defective in their neutralization capacity; however, cross-reactive antibodies incapable of virus neutralization are harnessed by the dengue viruses for ADE. If this outcome is dependent on an optimal ratio of antibody to pathogen, ADE is theoretically possible for any pathogen that can productively infect FcR- and complement receptor-bearing cells (2).
Non-neutralizing antibodies against the influenza virus were shown to be protective (41), most likely through FcR and complement-mediated pathways. However, in the case of dengue virus infection, cells that are involved in such protective pathways augment virus infection and are sources of disease-causing soluble factors (6). Differences between dengue and influenza viruses in this regard, might result in different OAS effects. Influenza viruses have not been shown to productively infect FcR-bearing immune cells or endothelial cells in humans (61). However, inflammatory responses originating from virus-antibody-complement-complement receptor complexes on pulmonary endothelial cells and other FcR- and complement receptor-bearing myeloid lineage cells (5762) cannot be ruled out. Conceptually, this situation could unfold anytime an optimal or protective ratio of antibody versus pathogen is disrupted by a rate increase in the number of pathogens that is higher than the rate of production of potentially protective cross-reactive or cognate antibodies (62).
We might suggest the possibility of 'bad' OAS in the 1918 H1N1 pandemic, and likely in some cases of the p2009 H1N1 outbreak, based on insight from 'bad' OAS with the dengue virus. Rapid virus replication could occur based on the unusually high virulence of the 1918 H1N1 virus (45). This would rapidly mobilize cross-reactive memory B cell responses in young adults, which were not sufficient for neutralization but still generated large amounts of immune complexes, with the rapidly replicating virus. This, in combination with the reaction with complement-complement receptors on pulmonary endothelial cells and myeloid lineage cells such as macrophages and neutrophils, could have resulted in massive inflammatory responses. Reported symptoms of infection with the 1918 influenza virus suggest that this scenario was very likely; autopsies and physician reports during the 1918 pandemic described symptoms of hemorrhagic tracheitis, bronchitis, and edematous hemorrhagic bronchopneumonia (53). This suggests that when an influenza virus replicates quickly, non-neutralizing cross-reactive antibodies generated from a memory response can potentially be more detrimental than the naïve immune reaction (62). However, it is unreasonable to expect an immunocompetent adult to not have any OAS against the influenza virus. Conceptually, the appearance of an unusually virulent, fast replicating influenza virus variant in a vaccine mismatch situation, could recapitulate disease severity seen in the 1918 pandemic in infected immunocompetent adults.
Go to :
CONCLUSION
OAS can be harnessed to defend against a pathogen, or a pathogen can harness OAS to gain entry into the host cell through the mechanism of ADE. OAS might also result in exaggerated inflammatory responses when there is disparity between the pathogen proliferation rate and the protective potential against the pathogen, which could be provided by cross-reactive memory B cells. The normally protective cross-reactive memory B cell responses of OAS being turned into 'self-destructive' modes of immune complex formation with a pathogen, during 'battle' against a fast replicating pathogen appears to be the inevitable casualties of the 'war' between the human immune system and pathogens. From an evolutionary point of view, MHC diversity in humans might represent a mobilization against such event; but from the individual point of view, young adults who have an OAS repertoire against influenza viruses, potentially through many natural infections and even without vaccination, could become casualties. The possibility of recapitulating the disease severity of the 1918 pandemic exists, even if on a micro scale, as the influenza virus circulates every season. Therefore, sustained surveillance for identifying an unusually virulent influenza virus variant should be continued.
Go to :
 XML Download
XML Download