

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Food allergy, the adverse immune response to foods, currently affects approximately 8% of children and 4% of adults. Its prevalence is rapidly increasing due to unknown reasons causing it to become a major public health concern in westernized and developed countries (12). Although hundreds of foods can trigger an allergic reaction, the big 8 foods, including milk, egg, peanut, soy, wheat, tree-nut, fish and shellfish, are responsible for most significant allergic reactions (3). Acute allergic reactions to food antigen range from mild to severe or life-threatening. Nearly 40% of food allergy patients visit emergency rooms at some point with severe or life-threatening food allergy responses characterized by hypotension, vascular collapse, and cardiac dysrhythmias, and over 30% have allergic response to multiple foods. Currently, the accepted standard of care for food allergy patients is strict avoidance of the diagnosed food allergen and injection of anti-histamines or epinephrine in case of accidental ingestion. However, these approaches negatively affect patient quality of life and adversely affect families who worry about accidental exposure and thus, limit their participation in social activities (45). Therefore, it is imperative that understanding of the immune responses to food allergens be expanded. Although food allergy is roughly divided into immediate IgE-mediated or delayed non-IgE-mediated allergic reactions, IgE-mediated food allergy is the most common and well defined of the two.
THE IMMUNE SYSTEM IN THE GASTROINTESTINAL MUCOSAL
The gastrointestinal (GI) tract is the largest interface between the body and the external environment. Although the main function of GI tract is to process ingested food into a form that can be absorbed, the GI tract also prevents microbes from invading. Because the intestinal mucosa is exposed to a huge quantity and diversity of foods and microbiota, it has the added challenge of distinguishing between harmful pathogens and beneficial commensal organisms (89). The GI tract has a large, well-organized mucosal immune system comprised of a three-layer defense. The first layer is a physical barrier comprised of epithelial cells including enteroendocrine cells, goblet cells, and paneth cells. Goblet cells and paneth cells establish major physical barrier by tight junction that is important for regulating intestinal permeability. A second defense layer is mucus. Goblet cells and paneth cells also can establish biochemical barrier by secreting mucus like mucin, anti-microbial peptide (AMP) and β-defensins. This mucus cover epithelial surface and block micro-organisms from reaching epithelial cells (101112). A third defense layer is densely populated immune cells that constitute either inductive sites such as peyer's patch (PP), mesenteric lymph nodes (MLN) and isolated lymphoid follicles or effector sites like lamina propria (LP). Antigen specific immune responses are induced in inductive sites and the migration of immune cells from inductive to effector sites is basis for cellular immune response in the GI tract. Lamina propria which lies below the epithelium is rich populated by many kinds of cells including fibro-blasts, CD4+ T cells, CD8+ T cells, regulatory T cells, plasma cells, dendritic cells, eosinophils, macrophages and mast cells. As the effector sites of the mucosal immune system, lamina propria recognize or combat invading pathogens. Thus, the epithelial cells, mucus layer and immune cells constitute the intestinal mucosal immune system and the interconnection or communication among 3 defense layer enables the mucosal immune system physically and immunologically to protection of the vast GI tract from invading microbes while maintaining commensal microbiota (13).
ADVERSE IMMUNE RESPONSE TO FOODS: FOOD ALLERGY/FOOD HYPERSENSITIVITY
Even though foods are non-self antigens, the immune system must somehow become non-responsive to food dietary antigens. The suppression of immune responses to previously orally-administered dietary antigens is known as "oral tolerance" (1415). Oral antigen administration in humans and mice has been shown to generate tolerance and induce a number of regulatory T cells. However, the breakdown or failure of oral tolerance to food antigens induces an adverse immune response known as "food allergy" or "food hypersensitivity" (16). Diverse, experimental food allergy mouse models have been developed in order to investigate the immune mechanisms underlying food allergy (17). In mice, food allergy can be developed by sensitizing with alum (18), by administration of oral antigen with a strong adjuvant like cholera toxin or staphylococcal enterotoxin (19), or by disruption of regulatory T cell function. These experimental food allergy models have generally shown that the immunological mechanisms involved in the development of food allergy consists of two phases 1) allergic sensitization, which is breakdown of oral tolerance, and 2) the effector phase, which is driven by effector cells reacting to the food allergen with results ranging from mild symptoms to severe anaphylaxis.
CLASSICAL IMMUNE MECHANISMS OF IgE-MEDIATED FOOD ALLERGY
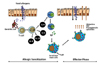
IgE-mediated food allergy is the most common and classical immune mechanism in food allergy and is mediated by the IgE/FcεR/mast cells axis in a two-step process consisting of sensitization and hypersensitivity reactions (Fig. 1). After ingestion, food proteins cross the epithelial barrier, make contact with the immune system of the GI tract, and are processed and presented by antigen-presenting cells. In the presence of IL-4, naïve CD4 T cells differentiate into Th2 cells and produce other type-2 cytokines like IL-5, IL-13, and IL-9 with IL-4 (20). These type-2 cytokines promote B cells differentiation into IgE-producing plasma cells (21) as well as intestinal mast cell proliferation and accumulation known as mastocytosis (22). Finally, allergen-specific IgE is distributed systemically and binds to the FcεR on mast cells and basophils (23). Together these processes constitute sensitization, poising the body to be reactive to food allergen. After sensitization, cross-linking of re-exposed food allergens to allergen-specific IgE that binds to FcεR on mast cells induces degranulation of mast cells (24) and release of several kinds of mediators such as histamine, platelet-activating factor (PAF), prostaglandin, and cytokines. This, in turn, causes hypersensitivity reactions ranging from a mild response to severe anaphylaxis (1825). Of note, the anaphylactic responses produced during the effector phase are induced quickly/immediately after ingestion of allergen-containing foods and are potentially life threatening (2627). Although Th2 cells, mast cells, and allergen-specific IgE all play major roles in classical IgE-mediated food allergy, the mechanisms that promote uncontrolled type 2 immune responses to dietary allergens in the GI tract and the underlying cause of the susceptibility to food allergy remain unclear.
THE ROLE OF IL-25 AND IL-25 RESPONDING CELLS IN IgE-MEDIATED FOOD ALLERGY
Multiple factors such as antigen availability, antigen uptake/presentation by dendritic cells (282930), costimulation (3132), function of regulatory T cells (3334), and the immune environment have been shown to contribute to the breakdown of oral tolerance and result in the induction of an allergic reaction to ingested food. Recently, emerging data have shown the importance of maintaining the intestinal epithelial barrier for prevention of GI allergic inflammation. As the primary barrier of the gut, the intestinal epithelium contains tight junctions that limit access of intact antigens and prevent them from encountering the immune system. For the induction of allergic responses, food proteins have to cross this intestinal epithelial barrier. Alteration of the intestinal epithelium by environment factors or stimuli such as inflammation or helminth infections are known to disrupt the integrity of the intestinal epithelium leading to increased intestine permeability and paracellular transport. In addition to this physical and physiological role, the intestinal epithelium also produces cytokines such as thymic stromal lymphopoietin (TSLP), IL-33, and IL-25 in response to barrier disruption by certain stimuli. These epithelial-derived cytokines have been shown to promote production of type 2 cytokines such as IL-5 and IL-13 from type 2 innate lymphoid cells (ILC2s) (353637) resulting in the initiation and amplification of type 2 immune responses in both allergic asthma and against parasitic infections (383940414243).
A cutaneous sensitization model of food allergy showed that TSLP enhances allergic sensitization and IL-33 is essential for inducing IgE-dependent anaphylaxis (44). In addition, IL-25 is also involved in the dysregulated type 2 immune responses to ingested food antigens and contributes to IgE-mediated food allergy susceptibility (6). In a food allergy mouse model that uses ovalbumin (OVA)/alum sensitization, repeated oral antigen challenge was shown to induce Il25 gene expression by the intestinal epithelium before onset of the anaphylactic response (18). Moreover, intestinal IL-25 transgenic mice (iIL-25Tg), which constitutively overexpress murine IL-25 in the epithelium of the small intestine, are more prone to IgE-mediated food allergy than their wild-type counterparts. Further, Il17rb–/– mice, which are deficient for the IL-17RB subunit of the IL-25 receptor, are more resistant to IgE-mediated food allergy. Together, these findings suggest that intestinal IL-25 signaling regulates susceptibility to IgE-mediated food allergy.
Two dominant IL-17RB-expressing cells, CD4+Th2 cells and ILC2s, have been identified in the lamina propria of the small intestine during development of experimental food allergy. Between CD4+Th2 cell and ILC2s, only intestinal ILC2s produce high amounts of IL-5 and IL-13 in response to IL-25 administered with IL-2 or IL-7. The direct contributions of ILC2s in promoting IgE-mediated food allergy have been demonstrated in Rora or Il17rb-deficient bone marrow reconstituted mice that are defective in ILC2 development or function, respectively. Moreover, IL-13-deficient bone marrow reconstituted mice are also resistance to food allergy, suggesting IL-13 production by ILC2s might also play a key role in promoting susceptibility to food allergy. This may be due to the induction of goblet cell hyperplasia, increase of intestinal permeability, or alteration of gut barrier function by IL-13 (4546). Furthermore, naïve or sensitized iIL-25Tg mice that have excess amounts of IL-25 signal and ILC2s do not drive allergic responses in mouse models. The capability of ILC2s to produce IL-5 and IL-13 in response to IL-25 and to promote experimental food allergy is enhanced in these mice due to the induction of increased numbers of IL-17RB+CD4+Th2 cells after repeated intragastric antigen challenge. Thus, IL-25 and IL-17RB+CD4+Th2 cells induced by ingested antigens enhance ILC2-derived IL-13 production, which may be a key step in amplifying the allergic reaction cascade to ingested antigens during the effector phase of IgE-mediated food allergy.
THE ROLE OF IL-9 AND IL-9 PRODUCING MUCOSAL MAST CELLS IN IgE-MEDIATED FOOD ALLERGY
The measurement of food allergen-specific IgE in serum is used as a diagnostic for food allergy as elevated allergen-specific IgE is indicative of sensitization. However, this is not a good parameter for prediction of symptoms or clinical reaction in food allergy patients. Indeed, among those with high levels of food allergen-specific serum IgE, only some individuals experience allergic responses following exposure. Thus, food allergen-specific IgE is required for the pathology of food allergy, but is not sufficient to induce disease. Therefore, there are additional, unidentified regulatory or signaling steps potentiating the susceptibility to food allergy.
The experimental food allergy mouse model using ovalbumin (OVA)/alum sensitization and repeated oral OVA challenge (18) has been reported to induce expansion of mucosal mast cells derived from bone marrow progenitors in the small intestine (47). As mentioned earlier, mast cells are well known effectors of allergic responses, releasing many mediators such as histamine and PAF and producing pro-inflammatory cytokines. Moreover, depletion of mucosal mast cells, inhibition of mast cell activation, and use of FcεRI-deficient mice all result in significant reduction of the development of allergic diarrhea following oral antigen challenge (18). Thus, mast cell activation is likely important for the elicitation of clinical reactions to oral food antigens. Recent studies have shown that IL-9 plays a role in promoting intestinal mastocytosis, characterized by the accumulation of mast cells in the intestine (2248). In the same ovalbumin (OVA)/alum sensitization model, sensitized IL-9- or IL-9R-deficient mice were found to be resistant to food allergy, exhibited failed mucosal mast cell expansion and mast cell degranulation, and reduced allergic diarrhea following oral OVA challenge, even though they produced OVA-specific IgE normally. On the other hand, intestinal-specific IL-9 overexpressing mice are more susceptible to food allergy (224849) and systemic IL-9 overexpression in mice also results in increased mast cell numbers (50).
In humans, microarray analysis identified IL-9 as the gene with the greatest difference in expression (among 12,257 differentially expressed genes) between children with peanut allergy and controls (51). Thus, IL-9 may also be required for clinical reaction to oral food antigens. Although it has been reported that Th9 cells (52), bone marrow (BM) - derived mast cells (53), eosinophils (54), neutrophils (55) and type-2 innate lymphoid cells (56) all produce IL-9, the major IL-9 producer in the intestine during food allergy is IL-9 producing mucosal mast cells (MMC9) (7). Recently, lineage-negative (lin–) IL-9-producing cells, named MMC9 after characterization, were identified in the lamina propria of sensitized and challenged mice. MMC9s expressing lin– c-Kit+ FcεRI+ST2+ β7 integrinlo are multifunctional and possess the unique characteristics of mucosal mast cells. In addition, MMC9s highly express mucosal mast cell-related chymase transcripts, Mcpt1 and Mcpt4, and the connective tissue mast cell-related tryptase transcripts, Mcpt6 and Gata1 and Gata2 with Il9. Moreover, these cells produce large amounts of IL-13 and IL-9 when stimulated with IL-33/SCF/IL-3 and release comparable amounts of histamine and MCP-1 as bone marrow-derived mast cells on ex-vivo IgE cross-linking. When cultured with SCF and IL-3, MMC9s rapidly develop into granular mast cells and acquire β-hexoaminidase activity while lost the capacity for IL-9 production. Interestingly, depletion of CD4 and use of genetically-modified mice such as IL-4-, IL-4R-, STAT6- or T cell-deficient mice significantly reduces MMC9s and mucosal mast cells and results in the failure to develop allergic diarrhea in response to oral antigen. Therefore, these finding suggest MMC9s are hypogranular mast cell lineage intermediates developed during type-2-cell-mediated intestinal inflammation.
The presence of MMC9s is positively correlated the degree of intestinal mastocytosis, mast cell activation, and allergic diarrhea. Further, in the absence of MMC9s, mice do not develop intestinal mastocytosis or food allergy symptoms. Interestingly, both IL-9 and IL9R-deficient mice develop MMC9s normally, but exhibit reduced numbers of mature mucosal mast cells and decreased mast cell activation after oral antigen challenge, indicating that the IL-9 is an important mediator of the differentiation of MMC9s into fully-mature mucosal mast cells. In addition, transfer of WT bone marrow, but not IL-9-deficient bone marrow, into irradiated IL-9-deficient mice restores the induction of intestinal mastocytosis and mast cell reactivity, indicating MMC9s generate IL-9 to facilitate their own maturation into mucosal mast cells. Moreover, MMC9s appear to regulate mast cell immunopathology in food allergy in that they can produce high amounts of IL-9 resulting in mastocytosis while they lose IL-9 production upon mucosal mast cell maturation. Thus, expansion of intestinal MMC9s might be a second signal required for clinical reactions associated with food allergy. Therefore, it has been speculated that MMC9s are responsible for the susceptibility to food allergy in effector phase.
CONCLUSIONS
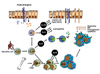
Here, we reviewed the immune mechanisms responsible for IgE-mediated food allergy in the gastrointestinal (GI) tract and in particular, the roles of IL-25 and ILC2s and of IL-9 and MMC9s (67). In addition to the role of allergen-specific Th2 cells in regulating food allergy, it has recently been described that IL-25, ILC2 cells and MMC9 cells also have key roles in amplifying the allergic reaction cascade in response to ingested antigens during the effector phase of IgE-mediated food allergy. Susceptibility to food allergic responses appears to be determined not by one parameter, but by a collaboration of several parameters that result in synergic responses. Indeed, the function of ILC2s and MMC9s in developing food allergies appears to also require CD4+Th2 cells. Moreover, the IL-25 mediated collaboration between ILC2s and CD4+Th2 cells potentiates the type 2 cytokine environment and physically changes intestinal permeability through IL-13 production by ILC2s. Moreover, the induction of mastocytosis by MMC9 cell expressed IL-9 and the interplay between MMC9 and CD4+Th2 cells appear to govern the susceptibility to and severity of food allergies (Fig. 2). Thus, understanding the factors required to initiate these events in response to dietary antigens might provide novel therapeutic strategies for the care of IgE-mediated food allergy patients.
 XML Download
XML Download