

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
STATs (Signal Transducer and Activator of Transcription) are latent cytoplasmic transcription factors that activate gene transcription in response to cytokines. In response to extracellular cytokine signals and growth factors, STAT proteins undergo tyrosine phosphorylation and are involved in many different regulatory events such as hematopoiesis, immunomodulation and development (1). Phosphorylated STAT6 is transported to the nucleus, where along with co-factors, STAT6 regulates the expression of genes that are involved in allergic inflammatory responses. Upon exposure to an environmental allergen, epithelial cells in the airways, esophagus and the keratinocytes in the skin secrete cytokines that initiate the development of an immune response (2). Chronic challenge with allergen results in organ-specific diseases such as asthma or food allergy that are characterized by pulmonary and gastrointestinal inflammation respectively.
In this review, we describe the role of STAT6 in the development of allergic disease with a focus on PARP14, a STAT6 transcriptional co-factor, and the related factor PARP1. Although neither PARP protein is required for T cell development, deficiency in either factor has effects on T helper cell differentiation and allergic inflammation. We review the role of STAT6 in allergic airway disease (AAD), atopic dermatitis (AD), food allergy and eosinophilic esophagitis (EoE) (Fig. 1). We then summarize research on the STAT6/PARP14 pathway and the cells where this pathway is important.
STAT6
Signal transducer and activator of transcription 6 (STAT6) is a member of the STAT family of proteins, which consists of STAT1, 2, 3, 4, 5A, 5B and 6. Human STAT6 is located on chromosome 12q13.3. The cytokines IL-4 and IL-13 activate STAT6 (3). The type I IL-4 receptor is composed of the common gamma chain (γc) and the IL4Rα chain and binds only IL-4. The type-II IL-4 receptor is composed of IL4Rα chain and IL13Rα1 chain and binds to and transmits signals of both IL-4 and IL-13. Three central tyrosine residues in IL4Rα chain (Y575, Y603 and Y631) become phosphorylated by stimulation of the receptor and are important for the activation of STAT6. These phosphotyrosines on the receptor chain become docking sites for SH2 domains of STAT6 monomers. The SH2 domains then get tyrosine phosphorylated themselves by receptor associated Jak kinases (Jak1 & 3). This allows for dimerization of STAT proteins via their SH2 domains. The resulting STAT6 dimer can now migrate to the nucleus where it can bind DNA directly (456).
Early studies in Stat6–/– mice showed that STAT6 is involved in IL-4 mediated functions including Th2 cell differentiation, class switching to IgE and expression of cell surface markers such as MHC-II and CD23 (789). STAT6 regulates the expression of Gata-3 and Gata-3 reconstitutes Th2 development in STAT6-deficient T cells, making it a critical factor for Th2 cell differentiation and function (10).
Asthma
Asthma is a chronic lung disease with intermittent recurring periods of coughing and wheezing caused by airway obstruction, abnormal lung physiology and airway hyperresponsiveness (AHR). It affects people of all ages but predominantly starts at childhood. Pathologic manifestations of asthma include airway inflammation, airway remodeling and increased secretion of mucus. Disease initiation and progression depends on Th2 cytokines IL-4, IL-5 and IL-13 and STAT6 activation is involved in IL-4/IL-13 mediated responses and development of inflammation in allergic airway disease (AAD). In an Ovalbumin-induced model of AAD, STAT6 deficient mice have greatly attenuated eosinophilia in BAL, airway hyperresponsiveness (AHR), mucus production and lung inflammation caused by the antigen (1112131415). Increased expression of IL-13 and IL-4 in mouse airway epithelial cells are able to reproduce the pathological conditions characteristic of asthma (1617). As STAT6 is responsible for the clinical manifestations of allergic asthma, many groups are studying it as a therapeutic target. STAT6 inhibitors such as STAT6-IP (Stat6 inhibitory protein) inhibit the Ova-induced development and secretion of Th2 cytokines, mucus production, lung inflammation and AHR (1819). These findings suggest that STAT6 plays a significant role in the development and progression of AAD.
Atopic dermatitis
Atopic dermatitis (AD) is a chronic, relapsing inflammatory condition of the skin. The key characteristics of AD include pruritus, scratching and chronic relapsing lesions. AD is characterized by increases in total IgE levels, infiltration of mast cells and eosinophils, decreases in anti-microbial peptides and increased expression of Th2 cytokines (20). The cause of AD is still unknown though genetic factors play an important role. Mutations in the epidermal differentiation complex (EDC, important during terminal differentiation of keratinocytes) genes such as filaggrin (FLG) are among those that can cause skin barrier impairment (2122). STAT6 and Th2 cytokines, IL-4 and IL-13 decrease the expression of EDC genes filaggrin (FLG), loricrin (LOR) and involucrin (IVL) in human skin biopsies and in differentiated primary human keratinocytes (2324). IL-4 and IL-13 are elevated in the skin of patients with AD and either cytokine can lead to decreased expression of antimicrobial genes like human b defensins (HBD)-2 and 3 (2025). Atopy is generally associated with increased activation of STAT6, hence a transgenic mouse model with constitutively phosphorylated STAT6 in T cells was generated to define a direct role for STAT6 in allergic inflammation (26). STAT6 transgenic mice expressing a constitutively active form of STAT6 in T cells have a hyper Th2 environment and are predisposed to develop allergic inflammation in the lung, skin and ocular tissue (272829). The absence of endogenous IL-4 protects the STAT6VT transgenic mice from allergic inflammation (2728). Transgenic mice expressing IL-4 or IL-13 in the epidermis of the skin developed chronic inflammatory pruritic skin disease, which is very similar to AD in humans (3031). Thus, STAT6 and Th2 cytokines along with other factors mediate skin barrier disruption and play a crucial role in atopic dermatitis.
Food allergy and eosinophilic esophagitis
As defined by the National Institutes of Allergy and Infectious Diseases (NIAID), food allergy is an abnormal immune response to food. The prevalence of food allergy appears to have increased over the last 2 decades, occurring more frequently in children than adults (32). Food allergy is more common in children with other allergic diseases and is most often IgE mediated. Reaction to food can result in skin manifestations, oral allergy syndrome, and can even result in anaphylaxis. Nut allergy is a common food allergy that has an early onset and is life threatening due to risk of anaphylactic shock. A single nucleotide polymorphism in the 3'UTR of the STAT6 gene is associated with susceptibility and severity to nut allergy in a UK population (33) and has been associated with food sensitization in a Mexican population (34). Anaphylactic response is usually the result from the activation of intestinal mast cells. IL-4 has an important role in IgE production and Il4-deficient mice do not develop experimental food allergy (3536). Studies have shown that IL-4 signaling has a direct effect on survival and proliferation of intestinal mast cells in food allergy (37). Along with IL-4, IL-9 also has an essential role in promoting intestinal mastocytosis and driving experimental food allergy (38). Studies in mice have shown the presence of multifunctional IL-9 producing mucosal mast cells that induce intestinal mastocytosis and increases in symptoms of food allergy (39). Increased IL-4Rα signaling confers increased intestinal permeability and sensitivity to food allergens (40). Oral tolerance to foods requires the development of allergen specific Treg cells and failure of Treg mediated oral tolerance is a major reason for food allergy. Allergen-specific Treg cells undergo Th2 cell reprogramming as a result of enhanced IL4R-STAT6 signaling which is sufficient to impart disease susceptibility (41).
Eosinophilic esophagitis (EoE) is a chronic inflammation of the esophagus and an emerging disease worldwide with a high rate of associated atopic disease (42). It is currently defined as a chronic, immune-mediated or antigen-mediated esophageal disease that is characterized by esophageal dysfunction and eosinophil-predominant inflammation. Children present with food impaction, dysphagia and have esophageal mucosal eosinophilia of >15 eosinophils per high power field (43). STAT6 mediates the expression of eotaxin-3/CCL26 (424445) and hence eosinophil accumulation in EoE (464748). Esophageal biopsies from children with EoE have an increased expression of CCL26 (49). Th2 cytokines IL-5 and IL-13 downregulate the expression of desmoglein-1 (DSG1) and genes of the epidermal differentiation complex (EDC) like filaggrin (FLG) (5051) and impair barrier function. Altered esophageal barrier could thus lead to enhanced antigen presentation and greater eosinophil recruitment. Taken together, these studies suggest an important role and function of STAT6 and Th2 cytokines in food antigen susceptibility and thus causing gastrointestinal inflammation such as food allergy and eosinophilic esophagitis.
MECHANISMS OF STAT6-DEPENDENT GENE REGULATION
The protein structure and domain organization of STAT6 includes the N-terminal domain that is involved in dimerization, the coiled-coil domain required for interaction with other proteins, the DNA binding domain, the SH2 domain (src homology 2 domain) that interacts with phosphorylated tyrosine residues in the receptor and the transactivation domain at the C-terminal end. STAT6 preferentially binds to a TTCN4GAA sequence and activates gene transcription. STAT6 recruits specific sets of co-activators to provide specificity for promoter activation (5). STAT6 recruits co-activators such as CBP/p300 and NCoAs to activate gene expression by providing histone acetyltrasnferase (HAT) activity. PARP-14 has been identified to be one such co-factor that associates with STAT6 and regulates gene expression.
PARPs
Poly (ADP–ribose) polymerases (PARPs) family of proteins contains 17 homologues, of which PARP1 is the founding member. PARPs are enzymes that catalyze the transfer of ADP ribose moieties from donor NAD+ molecules to target proteins although members of the family vary in their enzymatic abilities. Mono- and poly- ADP ribosylation is a protein modification that impacts the regulation of transcription, cell proliferation, apoptosis, signaling cascades and cell differentiation. Since the catalytic product of many of the members is unclear and some don't have a catalytic activity, a new nomenclature was proposed. Based on the type of enzymatic reaction and on structural features, PARP family members are now referred to as ADP-ribosyl transferase Diphtheria-toxin like (ARTD) family of proteins (52).
Of the 17 PARP family members, PARP1 is the most extensively studied. Reports using PARP inhibitors and knockout mice have shown that, in addition to its role in surveillance and maintaining genome integrity, PARP1 also functions in inflammatory diseases by triggering cell death in injured tissue (53). PARP family members including PARP1 and PARP2 are involved in T cell development in the thymus and in the periphery (5455). PARP1 regulates T cell activation and absence of PARP1 alters expression of genes and chemokines that are involved in maintaining Th1-Th2 balance (56). PARP1 and PARP14 have been studied to have a role in allergic inflammation.
PARP1 (ARTD1) and allergic inflammation: PARP1 was originally characterized for its role as a DNA damage sensor and its function in DNA repair. More recently, PARP1 has been identified for its role in transcription, chromatin structure modification, cell death pathways and in the mitotic apparatus among many others, thus having outcomes in carcinogenesis, genome maintenance, inflammation, neuronal function and aging (57). PARP1 participates in inflammation by directly or indirectly regulating the expression of inflammatory factors including cytokines, adhesion molecules and iNOS (58). Inflammation in the lung or any other organ causes an increase in PARP expression. PARP1 has emerged as a critical player in the development and the progression of allergic airway disease. Animal models of allergen (Ovalbumin) induced airway inflammation elucidates the positive involvement of PARP1 in airway inflammation, airway hyper responsiveness (AHR) (5960).
In an Ova triggered model of asthma, mice treated using pharmacological PARP inhibitors to target PARP1 and Parp1–/– mice had diminished AAD characterized by decreasing infiltration of inflammatory cells into the lung, reducing AHR and mucus secretion (59606162). This protective effect of absence of PARP1 is attributed to the inhibition of NF-kB activation and regulating the expression of molecules like TNF-α, iNOS and NFATc (5963). Decreases in the amount of IL-5 was linked to reduction in the number of eosinophils infiltrating the lung of Parp1–/– mice, suggesting that PARP1 regulates IL-5 production (58). The absence of PARP1 or its enzymatic activity severely decreases STAT6 protein without having an effect on STAT6 mRNA levels. STAT6 protein degradation was allergen stimulated and mediated by calpain as observed in an asthma model in Parp1–/– mice. This in turn results in reduced IL-5 levels, decreased eosinophilia and lung inflammation (64). Together, studies of PARP1 in models of asthma suggest that PARP1 directly regulates STAT6 and its absence can mitigate all signs of inflammation.
PARP14 (ARTD8) and allergic inflammation: PARP family members are classified into sub-families based on their domain architecture. One such sub-family is macro-PARPs that include PARP9, PARP14 and PARP15. Members of this family contain repeats of a domain found in the non-histone-like region of variant histone macroH2A (6566). PARP14 is enzymatically active and catalyzes mono-ADP ribosylation. Studies from many labs have shown that PARP14 functions in transcription regulation, signal transduction pathways and the actin cytoskeleton (67). PARP14 lacks the conventional transcriptional co-regulator domains such as HAT, Bromo, Chromo, SET or ATPase domains.
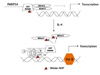
PARP14 associates with STAT6 in the presence or the absence of IL-4, but not with STAT1. The ability of PARP14 to promote transcriptional enhancement of IL-4 dependent and not IFNγ dependent gene activation lead to this factor initially being named CoaSt6 (for collaborator of STAT6) (Fig. 2) (68). Although the macro domains of histone macro H2A are known to participate in gene silencing, the macro domains of PARP14 interact with STAT6 and increase IL-4 induced gene expression (68). PARP14 and its associated PARP catalytic activity can mono-ADP-ribosylate itself and a STAT6 co-activator, p100 (Fig. 2). The enzymatic activity of PARP14 is required to enhance gene transcription and blocking PARP activity using pharmacological inhibitors blocked IL-4 dependent gene transcription in vivo (69). Studies show that PARP14 regulates STAT6-dependent gene transcription by functioning as a transcriptional switch. Under non-stimulating conditions, PARP14 binds to the STAT6-activated promoter and recruits HDAC2 and 3 to repress gene transcription. Upon IL-4 stimulation and STAT6 activation, the catalytic activity of PARP14 is increased resulting in ADP-ribosylation of itself and HDACs. The HDAC repressor complex leaves the promoter relieving gene repression and making the promoter more accessible to histone acetyl transferases (CBP/p300, NCoA-1 and NCoA-3) and increase gene transcription (Fig. 2) (70). As PARP14 functions as a transcriptional switch for STAT6 dependent gene transcription, naïve T cells from Parp14–/– mice cultured under Th2 conditions secrete reduced amounts of Th2 cytokines IL-4, IL-5 and IL-13 (71). Inhibiting PARP enzymatic activity using a pharmacological inhibitor confirmed a dose dependent reduction in Th2 cytokines, with similar amounts of IFNg produced from Th1 skewed cells (71). The mechanism for promoting Th2 differentiation is based on the ability of PARP14 or its enzymatic activity to regulate the binding of STAT6 to the Gata3 promoter. In a model of AAD, Parp14–/– mice have reduced numbers of inflammatory cells in BAL and reduced AHR.
Until recently, the role of PARP14 in other T helper cell subset differentiation and development was not known. Th17 cells are defined by their ability to produce IL-17A, IL-17F and IL-22 and to a lesser degree TNF and IL-6. They play a pivotal role in host defense and in induction and propagation of autoimmunity (72). Increases in IL-17A is correlated with increases in neutrophil recruitment and airway resistance in patients with asthma (73). T follicular helper (Tfh) cells are responsible for providing cognate help to B cells during formation of germinal centers. CD4 Tfh cells secrete IL-4, IL-10 and IL-21. IL-21 is required for germinal center formation for T-cell dependent antigens (74). Tfh cells are essential for IgE production and play an important role in allergic responses (75). ChIP-Seq analysis of WT and Parp14–/– Th2 cells identified that PARP14 along with its enzymatic activity regulated the expression of Il21 , and IL-21 promotes Th17 and Tfh development and function (76). It was also identified from the ChIP-Seq data that the expression of a number genes were dependent on the enzymatic activity of PARP-14 but were not regulated by STAT6, suggesting that PARP14 has STAT6 independent functions (76). Upon further studies, the absence of PARP14 or its enzymatic activity resulted in decreases in Th17 cells in the allergic airway inflammation model. PARP14 also promotes Tfh development by regulating the activation of STAT3, which is crucial for the differentiation of both Th17 and Tfh cell development (77). Similarly, Th17 and Tfh differentiation is diminished in PARP14 deficient T cells coincident with decreases in the phosphorylation of STAT3 (77).
PARP14 and its enzymatic activity also have functions in other cell types. In B-cells, PARP14 is required to maintain balance of B-cell subsets in the spleen and in generating IgA responses to antigen. PARP14 induces the expression of B-cell survival factors such as Pim-1 and Mcl-1 and protects IL-4 treated cells from apoptosis (78). PARP-14 deficient mice have significantly reduced numbers and frequency of Tfh cells. Tfh cells promote GC B cell development and a 45% reduction in the numbers and frequency of GC B cells was observed along with reductions in serum IgG1 and IgG2a/c titres in PARP14 deficient mice in response to SRBC immunizations. These could either be B-cell intrinsic effects or extrinsic mechanisms due to reduced Tfh numbers (77). Epithelial cells express PARP14 and this pathway has a significant role in airway epithelial cells where PARP14 deficient mice subjected to OVA induced AAD had diminished AHR (71). In esophageal epithelial cells, STAT6 regulates the expression of eotaxin-3 and it is found to be dramatically increased in patients with EoE (7980). PARP14 expression is increased in EoE biopsy samples with a positive correlation to eotaxin-3 expression, which was entirely dependent on STAT6 binding to eotaxin-3 promoter (49). Inhibiting the enzymatic activity of PARP14 in human esophageal epithelial cells cultured with IL4 and/or IL13, decreased the expression of eosinophil chemoattractant – eotaxin-3 (CCL26) (49). Keratinocytes, which are highly specialized epithelial cells also express PARP14. Our preliminary studies suggest that unlike its function in airway epithelial cells and esophageal epithelial cells, PARP14 does not impact expression of CCL26 or other epidermal differentiation genes (unpublished observations). These data together propose that PARP14 is expressed by a wide array of cells and STAT6/PARP14 pathway functions differently in specific cell types.
Contrasting the roles of PARP1 and PARP14 in allergic inflammation: As described in the previous sections, PARP1 and PARP14 work in distinct but overlapping pathways. In T cells, PARP1 has also been shown to modulate transcription factors such as STAT6, NFAT and NF-κB (5963), while PARP14 primarily regulates STAT6-dependent gene induction (70) and the activation of STAT3 (77). Although deficiency in PARP1 or PARP14 has similar outcomes in STAT6-mediated allergic inflammation, they regulate GATA3 gene expression and hence Th2 differentiation in very different ways. In the absence of PARP1, calpain mediates the degradation of STAT6 to reduce protein concentration within the cell (64), whereas in the absence of PARP14, HDACs remain bound to the Gata3 promoter, inhibiting gene expression (70). The absence of PARP14 and its enzymatic activity resulted in a reduced expression of IL-17A, IL-17F and IL-21 (77), whereas the absence of PARP1 has no effect on IL17 production (81). Lung cells of ovalbumin challenged Parp1–/– mice fail to synthesize GM-CSF and IL-5 resulting in impaired eosinophil recruitment (58). In comparison, the significant reduction in lung eosinophilia in Parp14–/– mice is attributed to the decrease in expression of CCL24 (71). Production of IL-4 was inhibited to a lesser extent than IL-5 and IL-13 following PARP-1 inhibition (58). Highlighting differences in mechanism, in clinical samples, there was a positive correlation between PARP14 and CCL26 expression in EoE patient biopsies, the same was not observed between PARP1 and CCL26 (49). Together, these studies suggest that PARP1 and PARP14 interact with different partners and regulate inflammation through independent functions.
CONCLUDING REMARKS
STAT6 plays varied functions in allergic inflammation both in immune cells and in resident tissue cells such as airway, esophageal, and intestinal epithelial cells, and keratinocytes (Fig. 1). Current therapies to treat allergic disease are targeted towards disease symptoms rather than the causative factor. Since STAT6 plays a pivotal role in allergic diseases, targeting the function of STAT6 has been an appealing therapeutic strategy. Along with STAT6, inhibiting the transcriptional co-activator PARP14 would be helpful. PARP inhibition might have beneficial effects in inflammatory conditions such as asthma (5862) and contact hypersensitivity (8283). In a mouse model of AAD, Parp14–/– mice have decreased lung inflammation, airway hyper responsiveness and serum IgE (71). Administering a pharmacological PARP inhibitor –PJ34 (that inhibits the enzymatic activity of PARPs) to wild-type mice that were subjected to AAD, reduced the severity of airway hyperesponsiveness and lung inflammation along with a reduction in the Th2 responses. Administering PJ34 either during challenge or sensitization and challenge or multiple times during the sensitization phase were effective (71). Recent studies show that PARP14 is required for STAT3 activation (77), suggesting that it could be a potential therapeutic target not only for allergic inflammation but also in other immune responses where this signaling pathway is important.

 XML Download
XML Download