

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Candida albicans is a major commensal fungus residing in the mucosal surfaces of our bodies. It is mainly found in the gastrointestinal tract, skin and genital tract. When the mucosal barrier is disturbed, C. albicans penetrates into the epithelium and causes systemic infection. Conditions associated with immunocomprimise, particularly neutropenia, are a major risk factor for invasive candidiasis (1). This deep-seated infection occurs, as hyphae spread to internal organs, among which the kidney is the major target (2). The mortality rate of patients with systemic candidiasis exceeds 30~40% even when potent antifungal agents are administered (3). Like many other pathogens, the host defense against invasive C. albicans infections is a function of immune detection and elimination of the pathogen (1). This defense strategy is referred to as resistance (4). However, mortality of the host following C. albicans infection may result largely from immunopathology, in which the immune response against C. albicans inflicts tissue damage. Mice are known to tolerate high fungal burdens, even though uncontrolled fungal proliferation is lethal. Thus, it is important to understand defense mechanisms that reduce host susceptibility to tissue damage caused by fungal infection or related immunopathologies, but that do not affect the fungal burden. This defense strategy does not affect fungal burden. In this review, we discuss the role of IL-33 in host resistance and tolerance to systemic C. albicans infections.
IL-33
IL-33 is a member of the IL-1 family of cytokines that delivers its signaling through ST2 receptors (5). Endothelial cells and epithelial cells rapidly produce IL-33 in response to a variety of intrinsic and environmental insults (6). This is why IL-33 is referred to as an alarmin. However, there are many sources of IL-33, and the kinetics of IL-33 production vary across types of cells and stimuli (7). Interestingly, the IL-33 receptor, ST2, is expressed on virtually all types of immune cells and numerous types of nonhematopoietic cells, suggesting that it exhibits a functional diversity (8). IL-33 function is context-dependent and can have opposing effects (e.g., inflammatory or anti-inflammatory) on an immune response. For example, IL-33 initiates type 2 immune responses through secretion of IL-4, IL-5 and/or IL-13 by Th2 cells and type 2 innate lymphoid cells (ILC2s) (9). IL-33 is also associated with autoimmune and inflammatory diseases mediated by Th1 and Th17 cells (6). In addition, anticancer and antiviral effects of IL-33 require CD8+ T cells, NK cells and ILC2s (10111213). Recent studies have identified anti-inflammation functions of IL-33: it plays a critical role in regulatory T cell expansion in acute colitis (14), asthma (15), and obesity (16). One effector molecule produced by regulatory T cells is amphiregulin, which plays a role in tissue repair without influencing antiviral immune responses (17). Thus, the seemingly anti-inflammatory effect of IL-33 is in fact a result of enhanced tolerance mechanisms. IL-33 also may exert its anti-inflammatory effects by suppressing Th1 or Th17-mediated immune responses. This outcome can be particularly important in inflammatory or infectious diseases in which Th1 or Th17 cells mediate immunopathology. Recent studies have begun to reveal the molecular mechanisms behind these observations. Specifically, IFN and IL-27 have been shown to counteract the activity of ILC2s mediated by IL-33 (181920).
IL-33 AND HOST RESISTANCE TO C. albicans INFECTIONS
The participation of IL-33 in host resistance to C. albicans infection was initially demonstrated in a fungal peritonitis model (21). IL-33 priming before infection with C. albicans resulted in effective fungal clearance and subsequently decreased mortality caused by fungal peritonitis (21). This priming by IL-33 acts on multiple steps of an anti-Candida neutrophil response. First, IL-33 priming enhances recruitment of neutrophils to the site of infection through two mechanisms: 1) It increases production of neutrophil-chemotactic CXCR2 ligands (CXCL1 and CXCL2) by peritoneal macrophages; and 2) it suppresses the internalization of surface CXCR2 in neutrophils. IL-33 does so by inhibiting TLR2-induced GRK2 expression that is required for CXCR2 internalization (22). Second, IL-33 priming of neutrophils increases their phagocytic and fungicidal activities. The increased phagocytosis of neutrophils occurs because of specific upregulation of the complement receptor CR3 through the synergistic activation of the TLR2 and Dectin-1 signaling pathways by IL-33. Increase in production of reactive oxygen species (ROS) is correlated with yeast killing of IL-33-primed neutrophils. It is not known why IL-33 specifically promotes clearance of opsonized C. albicans , but this occurred under physiological conditions (our unpublished data).
Mononuclear phagocytes, including resident macrophages and inflammatory monocytes, also play a critical role in Candida clearance (2324). Administration of IL-33 before and during C. albicans infection markedly induces M2 macrophage polarization (25). IL-33-induced M2 polarization is mediated by CD4+ T cell-derived IL-13 (25). Surprisingly, IL-33 stimulates bone marrow-derived M2 macrophages to efficiently phagocytize and kill C. albicans. The mechanism of action is not known, but one possibility is that IL-33 increases killing by directly upregulating Dectin-1 and mannose receptor or by indirectly inducting IL-13 production (2627).
Massive emergency hematopoiesis occurs after infections to satisfy the need for myeloid cells in the periphery. Under these conditions, hematopoiesis is preferentially directed toward active granulopoiesis and suppressed lymphopoiesis in the bone marrow. In addition, hematopoietic stem and progenitor cells (HSPCs) emigrate from the bone marrow to the periphery and active extramedullary hematopoeisis occurs. Injection of IL-33 induces massive expansion of myeloid cells, particularly eosinophis, in the periphery (5). Recently, we provided an explanation for how IL-33 mediates this phenomenon (28). Vascular endothelial cells produce high levels of CCL7 in response to IL-33, and CCL7, in turn, induces emigration of HSPCs out of the bone marrow with the help of CCR2. On the other hand, IL-33 directs the differentiation of HSPCs toward myeolopoiesis in both the bone marrow and periphery by an unknown mechanism. Systemic C. albicans infections induce tubular epithelial cells of the kidney to produce IL-33 (our unpublished data) and ST2 is highly expressed on HSPCs (8). Thus, it is tempting to propose that IL-33 may be involved in HSPC differentiation during C. albicans infection. This hypothesis is substantiated by the observation that C. albicans stimulates differentiation of HSPCs toward monocytes (29). Altogether, these results strongly suggest that IL-33 may control emergency granulopoiesis to supply sufficient numbers of granulocytes to the site of infection, thus, indirectly increasing host resistance to C. albicans infections.
IL-33 AND HOST TOLERANCE TO C. albicans INFECTIONS
Although administration of IL-33 increases host survival by promoting fungal clearance, the anti-inflammatory activity of IL-33 can also contribute to host survival during systemic C. albicans infections (25). There were significantly lower levels of proinflammatory mediators (IL-6, TNF-α, IL-1β, CXCL1, and CXCL2), relatively little neutrophil infiltrate, and M2 macrophage polarization in the kidneys of IL-33-primed mice throughout the course of infection. As mentioned previously, IL-13 mediates IL-33-induced M2 polarization. Interestingly, M2 polarization is required not only to keep fungal burden in check in the kidneys but also to maintain persistent immunosuppression associated with reduced Candida-induced immunopathology that leads to renal damage. Because hyperimmune responses are associated with more fatalities than high fungal burden in systemic C. albicans infections (29), IL-33 appears to enhance host tolerance to Candida infection via reduced immunopathology. Consistent with these results, IL-33–/– mice are more susceptible to systemic Candida infection compared to wild-type mice, showing impaired fungal clearance and severe renal immunopathology (our unpublished data). Severe renal inflammation is associated with a dysregulated differentiation of TNF-α- and iNOS-producing dendritic cells (Tip-DCs) in the kidney of Candida-infected IL-33–/– mice. In contrast, these mice have a lower number of IL-12- and IL-23-producing mature dendritic cells (DCs) in their infected kidneys. As reducing Tip-DCs decreases renal immunopathology without influencing fungal proliferation in IL-33–/– mice, IL-33 is a unique example of a factor that helps to reduce host vulnerability to damage by promoting pathogen clearance and reducing immunopathology (our unpublished data).
One important outstanding issue to explore is whether IL-33 is involved in types of tolerance other than immunopathology-related tolerance. IL-33 may control host tolerance to infections and other stresses through various means. For example, IL-33 can have direct protective effects on parenchymal cells of organs during a stress response (30), or IL-33 may exert tolerance through activation of cells that have a protective role against tissue injury. IL-33 administration expands ILC2s and regulatory T cells that can produce amphiregulin, a molecule critical for tissue repair and host tolerance (1731). Two other types of cells that can be targeted by IL-33 for tissue protection are M2 macrophages and Th17 cells. IL-22 secreted by these cells may be involved in tissue repair (3233). However, dysregulation of IL-33 can result in chronic inflammation or fibrosis (3435). Whether the tolerance mechanisms mentioned above operate in host defenses against systemic Candida infection needs to be tested. We propose that IL-33-induced tolerance is associated with an increased capacity for tissue repair and decreased immunopathology.
CONCLUDING REMARKS
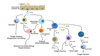
Mechanisms of IL-33 action in Candida immunity are beginning to be defined. IL-33 production reaches a peak in the kidney at day 3 of systemic C. albicans infection, when maximum inflammation and active fungal proliferation are occurring. At this time, IL-33 seems to prevent overactivation of an inflammatory response on one hand and to promote fungal clearance on the other hand (Fig. 1). The former process may result mainly from blocking inflammatory DCs, while the latter may occur at multiple stages of innate and adaptive immune responses to C. albicans infection. IL-33 can directly enhance the phagocytic and fungicidal activity of phagocytes. IL-33 also promotes IL-23 production by DCs which results in activation of the NK cell-GM-CSF-neutrophil axis. Finally, DC-derived IL-12 and IL-23 may be critical in IL-33-mediated fungal clearance through the induction of Th1 and Th17 differentiation, respectively. These beneficial activities of IL-33 have therapeutic implications for patients with invasive candidiasis. The ability of IL-33 to promote Th1 and Th17 responses against C. albicans also indicates that IL-33 has merits as an anti-Candida vaccine adjuvant.
 XML Download
XML Download