

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is a formidable health problem as it is the third leading cause of liver transplants in the United States and is predicted to surpass viral hepatitis and alcoholic cirrhosis as the leading cause in the next decade (1). NAFLD is characterized by extensive steatosis, or the accumulation of triglycerides in lipid droplets within hepatocytes. The etiology of NAFLD consists of complex interactions that often stem from obesity-related insulin resistance resulting in systemic dysregulation of glucose and lipid metabolism. Specifically, sedentary lifestyles combined with diets high in carbohydrates and saturated fats induce adipocyte dysfunction, resulting in increased uptake of free fatty acids by the liver. Hepatic steatosis is further exacerbated by the upregulation of de novo lipogenesis, which occurs despite a parallel rise in insulin resistance (2). A subset of patients is also predisposed to developing steatosis due to mutations in a number of genes, many of which regulate lipid metabolism (34). While simple steatosis is considered relatively benign, it can progress to non-alcoholic steatohepatitis (NASH), which is marked by the infiltration of immune cells into the liver. NASH can lead to the development of hepatic diseases such as fibrosis, cirrhosis, and hepatocellular carcinoma, and is also associated with an increased risk of cardiovascular disease (5). At present, treatment of NAFLD is largely limited to diet and lifestyle modifications while the gold standard for diagnosis is an invasive liver biopsy. Continued investigation of the molecular events that regulate disease progression are thus necessary to identify novel targets for the diagnosis and treatment of NAFLD.
The exact triggers that propel steatosis to NASH are not known; increased levels of free fatty acids, oxidative damage, hepatocyte death, and altered gut permeability can all contribute to the activation of local immune responses. Sustained activation of immune responses stimulates production of profibrogenic factors by hepatic stellate cells, initiating a tissue repair response that can manifest as end-stage liver diseases if left unchecked. Indeed, the contribution of the immune response to NAFLD progression is critical as deficiency or inhibition of innate or adaptive immune cells in mice results in less severe or no disease (67). In this review, we summarize recent advancements in our understanding of the roles of specific immune cells in the pathogenesis of NAFLD and discuss immunological factors that may serve as diagnostic or therapeutic targets.
HEPATIC IMMUNE RESPONSES
The liver is a unique immunological site as it is continually exposed to highly immunogenic content draining from the gut. This quasi-mucosal nature of the liver microenvironment requires homeostatic suppression of both immune cells resident in the liver and those in transit through the liver's sinusoids. The myeloid arm of liver-resident immune cells is enriched in macrophages and dendritic cells (DCs) that induce anergy in T lymphocytes, promote the generation of regulatory T cells (Tregs), or maintain hyporesponsive natural killer (NK) cells via secretion of immunosuppressive factors such as IL-10 (89). The lymphocytic compartment is enriched in NK cells as they comprise ~30% of lymphocytes in the human liver (10). Although hepatic NK cells can produce lytic agents and cytokines, including granzyme B, IFN-γ, and TNF-α, they also contribute to maintaining immune tolerance via fratricide of activated T cells (11). Similarly, another innate lymphocyte population in the liver, the natural killer T (NKT) cells, can limit T cell responses by inducing the upregulation of the inhibitory molecules PD-L1 and PD-L2 on hepatic antigen presenting cells (12). Moreover, interactions with T cells are not limited to hematopoietic cells, as they can be primed, albeit inefficiently, by hepatocytes, liver sinusoidal endothelial cells, and hepatic stellate cells (131415). Despite the multitude of mechanisms that dampen hepatic immunity, the liver retains the ability to stage robust immune responses upon inflammatory insult, such as injured hepatocytes in NASH, through recruitment of monocytes, granulocytes, and additional lymphocytes. Delineating the interplay among these populations will increase our understanding of the molecular switches that convert a quiescent immune environment into a cellular battlefield during chronic inflammatory diseases like NASH.
MONOCYTES AND MACROPHAGES
As the first line of defense against potentially pathogenic content draining from the gut, the liver must maintain a large population of phagocytic cells that can engulf and clear a diverse array of foreign compounds and bacterial products. It is perhaps for this reason that over 80% of the body's macrophages reside in the liver, including liver-resident Kupffer cells and their monocyte-derived counterparts, both of which play significant roles in the pathogenesis of NASH (16). Indeed, macrophage markers have been proposed as potential biomarkers of disease severity as levels of soluble CD163 and CD14 in the blood of NASH patients correlate with NAFLD activity score, degree of steatosis, fibrosis, and inflammation (1718).
Macrophages first appear in damaged adipose tissue, where they engulf dying adipocytes to form histological hallmarks called crown-like structures (19). Subsequent recruitment of monocytes to the liver is driven by chemotactic factors, such as monocyte chemotactic protein-1 (MCP-1). Production of MCP-1 is initiated by hepatocytes during simple steatosis and is sustained by infiltrating macrophages in a feedforward loop (720). As a result, blockade or absence of MCP-1 or CCR2, the receptor for MCP-1, reduces the influx of monocytes and macrophages into NASH livers, effectively halting the development of chronic inflammation (2122). Although MCP-1 is arguably the most well studied macrophage chemotactic factor in NASH, recent studies have identified additional molecules that regulate the influx of monocytes and macrophages into the liver, including the release of ATP or TRAIL from lipid-laden hepatocytes (2324). In addition, extracellular vesicles containing ceramide or CXCL10 released from injured hepatocytes are highly chemotactic to a number of immune cells, including macrophages (2526). Although these studies use bone marrow derived macrophages and cell lines to assess migration, their findings suggest that the trafficking of monocytes and macrophages to injured hepatocytes in NASH is a multifactorial process that can be targeted for therapeutic intervention. In fact, inhibition of CCR2 in NASH patients with fibrosis is an ongoing phase 2 clinical trial (NCT02217475). While such advances are promising, it will be interesting to determine whether the influx of monocytes occurs in parallel to in situ proliferation of liver-resident Kupffer cells and to further identify the relative contributions of each population toward the pathogenesis of NASH.
Upon infiltrating the liver during NASH, macrophages produce copious amounts of inflammatory cytokines such as TNF-α and IL-1β, which enhance steatosis and facilitate the development of fibrosis and hepatocellular carcinoma (72728). Pro-inflammatory cytokine production is triggered by imbalances in macrophage fatty acid and cholesterol metabolism or in response to damage associated molecular patterns released by injured hepatocytes (2930). These stimuli act in concert with toll-like receptors (TLRs), which are stimulated by elevated levels of endotoxin and other TLR ligands, as small intestinal bacterial overgrowth and loss of intestinal barrier integrity is characteristic of patients and animal models of disease (313233). Moreover, in a mouse model of NASH, Kupffer cells were hyperresponsive to low-levels of endotoxin, which was paralleled in blood monocytes of NAFLD patients (3435). These observations underscore a critical role for the gut-liver axis in NAFLD, wherein damage initiated in lipotoxic hepatocytes is converted to inflammation by recruited macrophages and further propagated by immunogenic products leaking from the intestine.
The pro-inflammatory nature of classically activated M1 macrophages that initiate NASH is in contrast to the anti-inflammatory phenotype of alternatively activated M2 macrophages that aid in the repair of damaged liver tissue. For instance, ablation of macrophages during liver fibrosis improves scarring; however, loss of macrophages during recovery from fibrosis increases scar formation, as macrophages are a critical source of collagenases that remodel fibrotic tissue (3637). In a carbon tetrachloride (CCl4) model of liver injury, restorative macrophages expressing some markers of both M1 and M2 macrophages infiltrate the liver at a late stage of disease, phagocytose dying cells, and resolve scar formation (38). Conversely, M2 macrophages induce proliferation and collagen production in fibroblasts and are strongly correlated with the expression of fibrogenic genes (39). Additionally, macrophages can directly contribute to fibrogenesis through collagen secretion (40). Crosstalk between M1 and M2 macrophages can also regulate inflammation as M2 macrophages induce apoptosis of M1 macrophages in a mouse model of alcoholic fatty liver disease (41). Nonetheless, in unresolved NASH, the restorative capacity of macrophages is perturbed given the decrease in their phagocytic ability, which correlates to the degree of steatosis (42). Promoting M2 macrophages or reducing M1 macrophages early in disease thus appear to ameliorate the progression of NASH, while sustained M2 skewing later in disease impairs effective wound healing responses and contributes to fibrogenesis.
DENDRITIC CELLS (DCS)
DCs are highly efficient antigen presenting cells that regulate immune responses through cytokine production and activation of T cells. Similar to macrophages, DCs play a dichotomous role in the pathogenesis of NASH and liver fibrosis. For instance, in a mouse model of NASH, DCs steadily accumulate in the liver in early stages of disease and produce significant amounts of the proinflammatory cytokines TNF-α, IL-6, and MCP-1 and the anti-inflammatory cytokine IL-10 (43). Surprisingly, depletion of DCs did not ameliorate disease and instead lead to increased hepatic infiltration of immune cells, elevated levels of pro-inflammatory cytokine production, notable loss in IL-10 production, and upregulation of fibrogenic markers (43). The beneficial effects of DCs in liver fibrosis are in part due to their ability to clear apoptotic debris and produce matrix metalloproteinases that enable clearance of fibrotic deposits (4344). Fibrosis is mediated by the activation and proliferation of hepatic stellate cells (HSCs), which are the pericytes of the liver that differentiate into myofibroblasts during fibrosis. In contrast to the net beneficial effect of DCs in fibrosis, culturing DCs from fibrotic livers with HSCs results in HSC proliferation and inflammatory cytokine production, suggesting that the effect of DCs in liver disease may vary by cell type (45).
Interestingly, DCs from fibrotic livers are able to induce robust cytolytic and proliferative antigen-specific T cell responses (45). On the contrary, DCs obtained from extrahepatic sites in high fat diet-fed mice are unable to initiate robust T cell responses (46). Although these studies differ in the models used, including a purely fibrosis model or diet-induced models of liver injury, they suggest that DC responses may be distinct between hepatic and extrahepatic sites in NASH and/or liver fibrosis. One explanation for these discrepancies may be intrinsic differences in lipid metabolism of liver-resident DCs compared to DCs in extrahepatic sites. Indeed, inhibiting global fatty acid synthesis resulted in ~20% loss of DCs from the spleen and bone marrow, while hepatic DCs were reduced by 80% (47). The increased sensitivity of hepatic DCs to changes in lipid metabolism may provide a potential therapeutic avenue, especially since DCs enriched in lipids are more immunogenic when compared to DCs with lower lipid content (48). Lastly, the distinct subsets of DCs in the liver could also be differentially modulated to alter local immune responses. A recent report of liver fibrosis following infection with Schistosoma mansoni found that a subset of DCs suppresses Th2 responses that lead to liver fibrosis (49). Moreover, DC depletion in a mouse model of NASH altered the ratio of CD8:CD4 T cells and reduced the intrahepatic frequency of regulatory T cells that dampen inflammation (43). Modulating DC responses may thus be an approach to limit T cell mediated liver injury (discussed below) in NASH.
NEUTROPHILS
Neutrophils are myeloid cells of the granulocytic lineage that are among the first cells to arrive at the site of inflammation. Unlike macrophages and T cells, neutrophils are not as prolific in the hepatic inflammatory infiltrate in NASH (50). Their muted presence may be reflective of the inflammatory mileu, as high-fat diet fed mice treated with LPS and IL-1β had an increased influx of mononuclear cells, while hepatic injury via carbohydrates and cholesterol stimulated the influx of both mononuclear and polymorphonuclear cells (51). Given the increased levels of endotoxins and metabolic insults in the liver during NASH, the resulting heterogeneity of the inflammatory infiltrate may thus mask the contribution of neutrophils to the progression of NASH. Nonetheless, a number of mouse models and neutrophil markers in NASH patients have helped identify effector mechanisms by which neutrophils contribute to the immunopathology of NASH. Specifically, expression of myeloperoxidase (MPO), an enzyme stored in the azurophilic granules of neutrophils that generates cytotoxic hypochlorous acid, is increased in the plasma and livers of NASH patients compared to patients with simple steatosis (52). MPO can oxidize phosphatidylcholine that further activates neutrophils and acts as a ligand for scavenger receptors, which in turn exacerbates fibrogenesis (53). Consequently, it is not surprising that deficiency of MPO results in a marked reduction in the production of pro-inflammatory cytokines and development of hepatic fibrosis (5254).
Release of MPO is part of the respiratory burst of activated neutrophils, which results in the production of reactive oxygen species (ROS). The importance of ROS in NASH pathogenesis is underscored by decreased circulating levels of antioxidants in obese children (55). Furthermore, neutrophils in the peripheral blood of NASH patients produce elevated levels of ROS upon stimulation when compared to controls (56). Local production of ROS can lead to lipid peroxidation and HSC migration, thus facilitating cellular injury and fibrosis (57). In addition to oxidative mechanisms of injury, neutrophils also secrete neutrophil elastase, which promotes hepatic insulin resistance by degrading insulin receptor substrate-1 (58). It is therefore conceivable that inhibitors of MPO, ROS, and neutrophil elastase may provide therapeutic benefit in NASH. Indeed, treatment of NASH patients with the antioxidant vitamin E reduced steatosis and serum levels of liver enzymes (59). Given the potential benefit in reducing neutrophil involvement during NASH, the increased ratio of neutrophils to lymphocytes in blood has been proposed as a potential noninvasive marker of disease severity (60). Although there is some debate as to the usefulness of this ratio in patients with comorbidites such as type 2 diabetes mellitus, further investigation of the role of neutrophils in the development of NASH may improve strategies for their use in diagnosis and treatment of disease (61).
NK CELLS
NK cells develop from common lymphoid progenitors, yet like many innate immune cells, respond to immunogenic insults early and in an antigen-independent manner. Mice lacking NK cells are resistant to developing steatosis following a high fructose diet, indicating that NK cells may have a role in facilitating the transition from NAFLD to NASH (6). Moreover, NK cells play a prominent role in chronic liver diseases, as they are critical players in inhibiting the development of fibrosis through direct killing of newly activated and senescent HSCs (6263). The increased cytolytic activity of NK cells is triggered by upregulation of activating stress ligands or downregulation of inhibitory ligands on HSCs (6364). However, HSCs that are "fully activated" or fail to become senescent are resistant to killing by NK cells (6263). In contrast, senescent HSCs downregulate fibrogenic programs and upregulate inflammatory genes (65). Considering that HSCs are chronically activated in the proinflammatory environment of the liver during NASH, these findings may be indicative of dysregulated senescence of HSCs in NASH. Yoshimoto, et al. demonstrate that induction of the senescence phenotype in HSCs is associated with increased rates of hepatocellular carcinoma in mice exposed to a carcinogen that were fed a high-fat diet compared to normal diet fed mice (66). Given that these experiments were conducted in mice with intact immune responses, these data may be indicative of defective NK cell function in NAFLD, as NK cells kill senescent HSCs.
We recently reported that depletion of hepatic NK cells expressing the activating marker NKp46 promotes fibrogenesis by skewing macrophages toward M2 phenotypes late in disease (67). The importance of NKp46 expression in regulating HSC activity was previously demonstrated in fibrosis resulting from viral hepatitis, as loss of robust NKp46 expression was inversely related to fibrosis grade (68). Moreover, blockade of NKp46 results in decreased NK cell degranulation, IFN-γ production, and HSC killing upon in vitro co-culture with HSCs (68). In contrast, total NK cells and expression of the NK cell activating receptor NKG2D and its ligand MIC A/B are elevated in NASH patients and are associated with increased fibrosis grade (69). While the authors of this study interpret the data as indicative of a potential pathogenic role for NKG2D-MIC A/B interaction in NASH, their findings could also reflect a compensatory, yet ineffective, increase in NK cell activity. This alternative interpretation further implies that NK cell responses may be compromised in NASH, thus permitting uncontrolled HSC proliferation and activation. Indeed, in a CCl4 model of liver fibrosis, production of IFN-γ by NK cells was diminished in advanced stages of disease (70). Interestingly, the reduction in NK cell IFN-γ production was partly alleviated by blocking TGF-β released from HSCs (70). Continued investigation is thus required to thoroughly understand the complex interactions among NK cells, HSCs, macrophages, and likely other immune and nonimmune cells in NASH.
NATURAL KILLER T (NKT) CELLS
NKT cells are defined by the co-expression of an invariant T cell receptor and NK cell markers and recognize lipid antigens presented by the non-classical antigen presenting molecule CD1 (71). Classical CD1d reactive hepatic NKT cells are differentially distributed in mice and humans, where Vα14Jα18 NKT cells constitute ~22% of hepatic mononuclear cells in mice, while the human homologue Vα24Vβ11 NKT cells comprise only ~0.6% of CD3+ cells that bind CD1d tetramers (7273). However, the counterpart to mouse invariant NKTs may instead be human mucosal associated invariant T (MAIT) cells, which make up ~15% of intrasinusoidal lymphocytes in humans (74). Given the differences between NKT cells in mice and humans, translating data obtained from mouse models of NASH to human disease may require caution.
NKT cell accumulation during NAFLD and NASH has been reported by several studies in both the liver and blood of patients (7576). Yet, other studies report that hepatic NKT cells are decreased in NASH patients and associate decreased frequencies of NKT cells in the peripheral blood with an increased risk of NAFLD (7778). These differences may in part reflect when and how NKT cells were detected. For instance, in a mouse model of NASH, NKT cells were selectively increased in the liver, but were unchanged in the spleen (79). However, the same study reported that NKT cells that infiltrate steatotic livers undergo apoptosis (79). Identifying the kinetics of NKT cell infiltration into the liver during the progression of disease in NASH patients may provide further insight into the frequency of NKT cells in NAFLD.
Influx of NKT cells into the liver during NASH is mediated by enhanced expression of the chemokine CXCL16 on endothelial cells and macrophages, which binds CXCR6 on the surface of NKT cells (80). Upregulation of CXCL16 coincides with the production of IL-4 and IFN-γ by NKT cells, which aggravates inflammation via macrophage activation (80). Activation of the Hedgehog pathway has also been implicated in NKT cell recruitment in NASH livers. Specifically, overactivation of Hedgehog signaling recruits increased numbers of NKT cells to the livers of methionine cholinedeficient diet-fed mice, which developed higher grade fibrosis compared to wild type mice (75). CD1d–/– mice lacking NKT cells did not develop fibrosis, suggesting that Hedgehog mediated NKT cell responses are pathogenic in NASH (75). Indeed, activation of the Hedgehog pathway in NKT cells upregulates expression of osteopontin production, which drives HSC activation and fibrogenesis (81). Thus, the net contribution of NKT cells appears to be pathogenic in NASH. Nonetheless, modulating the ability of NKT cells to produce cytokines may permit tuning of macrophage responses to favor the resolution of inflammation and clearance of fibrotic tissue.
T CELLS
T cells are a diverse class of lymphocytes that include CD4 and CD8 T cells, which respond to antigens displayed on MHCII and MHCI, respectively, to exert effector functions such as cytokine production and cytolysis. T cells play a critical role in the development and progression of NAFLD as high fructose-diet fed mice lacking T cells fail to develop steatosis and hepatic inflammation (6). These findings are validated in NASH patients as they have increased frequencies of IFN-γ+ memory CD4 and CD8 T cells (56). Elevated peripheral blood T cells in NASH patients are reflective of cells infiltrating the inflamed liver (82). One of the molecular mechanisms driving T cell infiltration into the liver is dysfunctional chemotaxis, as peripheral CD4 T cells from obese mice and NASH patients migrate more readily toward the chemokine CXCL12 when compared to T cells from healthy mice or donors (83). These results suggest that imbalances in systemic lipid metabolism may result in intrinsic alterations in immune cells. In addition, increased oxidative stress in the hepatic microenvironment of NASH livers generates neoantigens that can induce the recruitment of T cells (84). Collectively, these studies identify T cells as a prominent immune population in NASH that plays a critical role in influencing the course of disease.
Functionally, CD4 T cell responses in NASH are skewed toward Th1 and Th17 phenotypes. Th1 responses in NASH are characterized by secretion of IFN-γ and TNF-α, which in turn help polarize macrophages toward M1 responses (568284). Although the lack of steatosis and inflammation in the absence of T cells would suggest that T cell derived IFN-γ is pathogenic in NASH, there are no studies to date that detail the progression of disease upon neutralization of IFN-γ (6). In contrast, IL-17 is well known to propagate NASH via multiple mechanisms, including neutrophil activation. Moreover, exposure of HepG2 cells to fatty acids in the presence of recombinant IL-17 promotes accumulation of intracellular triglycerides (85). Additionally, IL-17 signaling in HSCs upregulates expression of profibrotic genes while lack of IL-17 in a chemically-induced murine model of liver fibrosis reduces the levels of proinflamamtory cytokines and extent of cell death (8687). Interestingly, fibrosis was exacerbated in mice lacking expression of IL-22, a cytokine produced by Th17 cells that promotes epithelial regeneration (87). Although the above studies provide ample evidence for a pathogenic role of Th17 cells in NASH, perhaps tuning Th17 responses to increase IL-22 production may uncover a beneficial role for the increased presence of this population in NASH livers.
The marked increase in Th1 and Th17 responses in NASH is complemented by a loss in regulatory T cells (Treg) in the adipose tissues, peripheral blood, or livers of NASH patients or mice that replicate facets of the disease (888990). In the liver, the reduced presence of Tregs is not only relative to the influx of other T cell subsets, but may also be due to increased susceptibility to death by oxidative damage (89). Importantly, adoptive transfer of Tregs into high-fat diet mice decreases hepatic inflammation and serum levels of liver enzymes, indicating that Tregs may regulate the transition from NAFLD to NASH (89). These findings were corroborated by the association of elevated hepatic lipogenesis and inflammation and reduced Treg frequency in mice lacking expression of the costimulatory molecules CD80 and CD86 (91). Interestingly, blockade of CD80 and CD86 in mice with an intact Treg compartment had the opposite effect as the grade of steatosis and fibrosis was decreased with a concurrent improvement in glucose tolerance (91). These contrasting results indicate that modulating costimulatory molecules in the presence of Tregs may be an attractive means to regulate T cell responses in NASH. Indeed, blockade or genetic deficiency of another costimulatory molecule, CD40L, impeded inflammation in both adipose tissues and livers of mice fed obesogenic diets (9293). Surprisingly, deficiency of CD40, which binds CD40L, produces the opposite results, as steatosis and insulin resistance were exacerbated in CD40–/– mice compared to wild type controls despite reduced hepatic levels of inflammatory cytokines (94). CD40L can also bind the integrins α5β1 and Mac-1 (9596). The contrasting progression of disease in CD40 and CD40L deficient mice may thus reflect interactions that are independent of CD40 and T cells as α5β1 and Mac-1 are expressed on several cells of nonlymphocytic origin.
EMERGING FRONTIERS
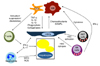
The immune response accelerates and magnifies the extent of injury, yet paradoxically facilitates the resolution of inflammation and fibrosis in NASH (Fig. 1). As discussed above, these multifaceted roles of the immune response offer several potential diagnostic and therapeutic targets for NASH. The benefit of these emerging targets may not only include controlling inflammation, but may also help reduce the dependence on existing pharmacological treatments for NASH. As an example, glitazones, which improve insulin sensitivity and enhance production of the adipokine adiponectin, are associated with an increased risk of cardiovascular dysfunction and bladder cancer (97). Perhaps combining immunomodulatory agents with existing treatment modalities may reduce net toxicity, while having the added advantage of combating both the cause and the effect of the disease.
Nonetheless, continued investigation of the interplay between various immune compartments is necessary to arrive at a complete understanding of the immunopathogenesis of NASH. As an example, IL-22, which protects against fibrogenesis, is produced not only by Th17 cells, but also by a subset of innate lymphoid cells (ILCs). Exogenous administration of IL-22 was reported to induce a remarkable reversal of insulin resistance, weight gain, and bacterial dissemination to the liver in obese mice (98). Furthermore, another subset of ILCs has been shown to regulate the conversion of white adipose tissue to brown adipose tissue, which protects against the development of obesity (99100). These exciting findings add ILCs as the newest player in the many complexities of hepatic immune responses; they remind us that the next frontier may bring the marker, cytokine, or cell type that can be targeted for the successful management of NASH and other chronic liver diseases.
 XML Download
XML Download