

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Expression of CD137 ligand (CD137L; also known as 4-1BBL and TNFSF9) is found mainly on professional antigen-presenting cells (APCs) such as dendritic cells, monocytes/macrophages, and B cells, and its expression is upregulated during activation of these cells (12). However, its expression has been documented on a variety of hematopoietic cells and nonhematopoietic cells (3). Generally, CD137L is constitutively expressed on many types of cells but its expression levels are low except for a few types of cells. Interestingly, CD137L is coexpressed with CD137 (also known as 4-1BB and TNFRSF9) on various types of cells (13), but expression of CD137 potently downregulates that of CD137L by cis-interactions between the two molecules resulting in endocytosis of CD137L (45).
CD137L signaling has been relatively well characterized in the process of inflammation, hematopoiesis, and immune tolerance. It is becoming clear that CD137L signaling is critical in multiple steps during the progression of inflammation (3). In most inflammatory conditions that are caused by various stresses, early events mediated by CD137L signaling may occur in vessels. For example, CD137 on activated endothelial cells stimulates CD137L on monocytes to enhance their extravasation through activation of cell adhesion molecules (6). During interactions between monocytes and endothelial cells, CD137/CD137L bidirectional signals are delivered into endothelial cells but only CD137L signaling takes place in monocytes. As a result, endothelial cells and monocytes secrete proinflammatory cytokines and chemokines (78). Once monocytes arrive at inflammatory sites, they are likely to differentiate into macrophages or dendritic cells depending upon the microenvironment of inflammatory sites. At the early phase of tissue inflammation when proinflammatory mediators become enriched in inflamed sites, CD137L signaling may elicit the differentiation of monocytes toward M1 macrophages or dendritic cells and amplify tissue inflammation. On the other hand, CD137L signaling in parenchymal cells also can contribute to a vicious cycle of tissue inflammation. For instance, CD137L signaling in tubular epithelial cells is a key point for exacerbating ischemia-reperfusion-induced renal inflammation by promoting recruitment of neutrophils into the kidney (910). These observations implicate CD137L signaling as a convergence point to amplify tissue inflammation through recruitment of inflammatory cells and increment of proinflammatory mediator production. It remains to be clarified whether CD137L signaling also functions as a key player after acute inflammation subsides. During the resolution phase of inflammation, however, it is likely that CD137L signaling help tissue repair mechanisms presumably by inducing M2 macrophage differentiation and regeneration of parenchymal cells including epithelial cells. It is needed to be noted that outcomes of CD137L signaling are context-dependent in most cases. Here, I propose that CD137L signaling functions as a costimulatory signal during inflammation: i.e., a primary signal determines the general fate or propensity of immune responses and the secondary CD137L signal reinforces the primary signaling. Primary signals are transduced via pattern recognition receptors (PRR) such as Toll-like receptors (TLR), C-type lectin receptors (NLR), NOD-like receptors (NLR) and RIG-I-like receptors (RLR), and receptors for a variety of proinflammatory mediators and growth factors. During inflammation, ligands for the primary receptors include pathogen- or damage-associated molecular patterns (PAMP or DAMP), inflammatory mediators, and growth factors. They may be provided by pathogens, damaged neighbor cells, or CD137-expressing CD137L counterpart cells. Our unpublished data demonstrated that Candida albicans activates macrophages through the primary Dectin-1 or TLR2 receptor and the secondary CD137L. Fulminant inflammation occurs after infection with C. albicans only when both signals are present in macrophages, and each signal cannot compensate for the other in the production of inflammatory mediators by macrophages. This two signal model implicates CD137L signaling as a safeguard that regulates the powerful and potentially harmful immune reaction and prevents the accidental triggering of responses against the host's own tissues (11).
Emerging results provide evidence that CD137L signaling is involved in emergency hematopoiesis (12). In particular, systemic infections require a great number of phagocytes such as granulocytes and macrophages. In this situation, a balance between myelopoiesis and lymphopoiesis is skewed towards myelopoiesis. In the steady state, CD137L signaling preferentially inhibits myelopoiesis and B cell generation (1314). Seemingly paradoxically, however, myelopoiesis is impaired in the bone marrow during infections in the absence of CD137L signaling (12). In some in vitro experiments, it has been shown that ligation of CD137L stimulates hematpoietic progenitor cells to differentiate into myeloid cells (1516). I will provide a plausible explanation for this discrepancy below.
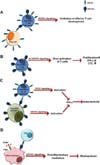
The results reported in the literature to date have shown, without exception, that CD137L signaling plays a proinflammatory role in inflammatory disease models (2917). However, recently, Croft and colleagues have demonstrated that CD137L signaling can restrain effector T cell development in a condition that antigen presentation is limiting (5). Delivery of CD137L signals is distinguishable in T cells when presentation of a low dose of antigen induces low levels of CD137 expression on T cells, since the number of CD137 molecules in the cell surface is not sufficient to internalize all of the surface CD137L molecules. Their findings are intriguing, as they may provide an insight into the dysregulation of immune responses that are often observed in CD137-/- and CD137L-/- mice. First, CD137L signaling may function as a brake for T cell activation (Fig. 1A). There are many reports showing that CD137-/- mice have dysregulated immune responses presumably due to lack in CD137L signaling in T cells (18-20). According to our unpublished results, over-activated CD8+ T cells in tumors of CD137-/- mice secrete a high quantity of IFN-γ, a cytokine required for M1 tumor-associated macrophage (TAM) differentiation (Fig. 1B). Once M1 TAMs are established in tumors, they and activated CD8+ T cells may activate each other, resulting in breaking of tumor immunological tolerance. Thus, they act synergistically on the enhancement of antitumor activity in CD137-/- mice. As wild-type regulatory T (Treg) cells abrogate the increased antitumor activity in CD137-/- mice, it is believed that Treg cells control the activity of CD8+ T cells, including IFN-γ production, via CD137-CD137L interactions. Second, CD137-/- mice are likely to have dysregulated immune responses when CD137-expressing cells are infused, because CD137L expression remains high in T cells and APCs of CD137-/- mice after their activation. For example, alloreactive T cells transferred to CD137-/- mice express high levels of CD137 on the cell surface after allostimulation. CD137 on alloreactive T cells in turn can activate host APCs via elevated levels of CD137L (due to the absence of CD137) and induce production of proinflammatory mediators, cell adhesion molecules, and costimulatory molecules (Fig. 1C). Lack in the CD137L downregulation mechanism by CD137 in CD137-/- APCs is believed to render them more sustainable and potent in stimulating alloreactive donor T cells (Fig. 1C). We frequently observe that transfer of alloreactive T cells breaks host immunological tolerance in CD137-/- mice, inducing severe autoimmune disease (our unpublished data). Finally, CD137-/- and CD137L-/- mice often have hypo-inflammatory responses to infections and other inflammatory stimuli. This may be caused directly by deficiency of CD137L signaling that is connected to the production of proinflammatory mediators. As mentioned above, macrophages cannot produce high levels of proinflammatory mediators in response to PAMPs in the absence of CD137L costimulatory signals (Fig. 1D). As proinflammatory mediators are indispensable for myelopoiesis during infections, the primary reason for impairment in myelopoiesis of CD137-/- and CD137L-/- mice can be attributed to hypo-inflammatory responses in these mice (Fig. 1D). By contrast, CD137L signaling potently accelerates myeloid cell differentiation in the presence of sufficient amounts of inflammatory mediators.
Taken together, CD137L signaling is likely to fine-tune immune responses in two ways. First, CD137L signaling controls a balance between immunological suppression and activation by restraining effector T cell development under tolerogenic and immunologically stimulating environments. Second, CD137L signaling can be regulated by induction of high levels of CD137 expression which can shut down CD137L signaling through endocytosis of CD137L in the same cell. Future studies will be guaranteed to prove these hypotheses.
 XML Download
XML Download