

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Osteoarthritis (OA), the most common form of arthritis, is a complex joint disease that produces debilitating chronic pain in affecting individuals. OA is characterized by progressive cartilage loss, osteophyte formation, and changes of subchondral bone. OA tends to worsen over time as cartilage wears away. However, there are increasing evidences supporting that inflammation may be implicated in the development and progression of OA, even in the early stage (1). Proinflammatory cytokines such as interleukin-1β (IL-1β) and IL-6 play critical roles in disturbed homeostasis in OA cartilage (2,3). Until now, treatment targets have mainly focused on reducing pain. Relatively, disease modifying anti-OA drugs (DMOADs) are still lacking.
Oxidative stress implies a disturbed cell state in which reactive oxygen species (ROS) production exceeds the neutralization capacity. Chronic oxidative stress underlies the pathophysiology of several autoimmune diseases including rheumatoid arthritis (RA) and Behçet's disease (4,5). Numerous studies have demonstrated that oxidative stress has been implicated in OA similar to RA, albeit to a lesser degree (6). In conjunction with metalloproteinases, ROS can work to degrade matrix components. ROS directly oxidizes transcriptional factors, intracellular and extracellular components resulting in cell death and matrix components breakdown (7-9). Previous reports have shown that ROS scavengers can attenuate the degradation of articular cartilage (10).
Ursodeoxycholic acid (UDCA) is a naturally occurring dihydroxy hydrophilic bile acid that is currently used to treat various liver diseases including primary biliary cirrhosis (11). Previous reports suggested that immunomodulation and anti-inflammatory effects are implicated in the mechanisms of the drug (12). And, the action of antioxidant of UDCA may contribute to its hepatoprotection properties (13-15). To our knowledge, the effect of UDCA has not been investigated in an animal model of OA or human OA. Given the potential role of oxidative stress in OA pathogenesis, the effects of UDCA on pain production and cartilage degeneration in a monosodium iodoacetate (MIA)-induced OA rat model was investigated.
MATERIALS AND METHODS
Animals
Male Wistar rats weighing 140~230 g (6 weeks of age) at the start of the experiment were purchased from Central Lab. Animal Inc. (Seoul, South Korea). The animals were housed three per cage in a room with controlled temperature conditions (21~22℃), controlled lighting (12-h light/12-h dark cycle) and access to sterile food and water. All animal procedures were approved by the Animal Research Ethics Committee at the Catholic University of Korea.
Induction of OA in the rat and treatment with UDCA
The animals were randomized and assigned to treatment groups prior to the start of the study. After anesthetization with isoflurane, rats were injected with 50 ml containing 3 mg of MIA (Sigma) using a 26.5-G needle inserted through the patellar ligament into the intra-articular space of the right knee. Control rats were injected with an equivalent volume of saline. UDCA was kindly provided by Daewoong Pharmaceutical Company (Seoul, Korea). UDCA dissolved in saline was administered orally every day. The vehicle-treated animals were given an equivalent volume of cotton seed oil solution. The dose of UDCA was 50 mg/kg of body weight.
Assessment of pain behavior
The MIA-treated rats were randomized to each experimental group. Nociceptive testing was performed using a dynamic plantar esthesiometer (Ugo Basile), an automated version of the von Frey hair assessment procedure, before the MIA injection (Day 0) and on the given day after MIA injection. The rats were placed on a metal mesh surface in an acrylic chamber in a temperature-controlled room (21~22℃) and allowed to rest for 15 min before testing. The touch stimulator unit was oriented beneath the animal. An adjustable angled mirror was used to place the stimulating microfilament (0.5-mm diameter) below the plantar surface of the hind paw. When the instrument was activated, a fine plastic monofilament advanced at a constant speed and touched the paw in the proximal metatarsal region. The filament exerted a gradual increasing force on the plantar surface, starting below the threshold of detection and increasing until the stimulus became painful indicated by the removal of its paw. The force required to elicit a paw withdrawal reflex was recorded automatically and measured in g. A maximum force of 50 g and a ramp speed of 20 s were used for all esthesiometry tests. Pain behavioral tests of secondary tactile allodynia were conducted prior to the UDCA administration.
Histological and immunohistochemical analyses
Histological changes were assessed to confirm the effect of UDCA on cartilage degeneration in the knee joints of OA rats. The animals were perfused via the ascending aorta with 10% neutral buffered formalin (pH 7.4). The knee joints, including the patella and joint capsule, were resected and kept in the same fixative for an additional 48 h at 4℃. The fixed specimens were decalcified with 5% formic acid decalcifier for 6 days at 4℃. After decalcification, the specimens were embedded in paraffin. Standardized 7µm serial sections were obtained at the medial and lateral midcondylar level in the sagittal plane and were stained with hematoxylin and eosin (HE), Safranin O-fast green, and toluidine blue to enable evaluation of proteoglycan content. Slides for immunohistochemistry were deparaffinized and rehydrated using a graded ethanol series. The sections were depleted of endogenous peroxidase activity by adding methanolic H2O2 and then blocked with normal goat serum for 30 min. The samples were incubated overnight at 4℃ with antibodies to IL-1β at a dilution of 1 : 50 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), matrixmetalloproteinase-13 (MMP-13) at 1 : 50 (Abcam, Cambridge, UK), IL-6 at 1 : 50 (Abcam), inducible nitric oxide synthase (iNOS) at 1 : 100 (Abcam), nitrotyrosine 1 : 100 (Santa Cruz). The samples were then incubated with the respective secondary antibodies, biotinylated anti-mouse IgG or rabbit IgG, for 20 min, conjugated to a streptavidine peroxidase complex (Vector Laboratories, Burlingame, CA, USA) for 1 h, and finally with 3,30-diaminobenzidine (Dako, Glostrup, Denmark). The sections were counterstained with Mayer's hematoxylin and photographed using an Olympus photomicroscope (Olympus, Tokyo, Japan). A modified Mankin's histological score was used to score histological injuries of the articular cartilage as follows. The structure was scored on a scale of 0~6, where 0=normal; 1=irregular surface, including fissures into the radial layer; 2=pannus; 3=absence of superficial cartilage layers; 4=slight disorganization (cellular row absent, some small superficial clusters); 5=fissure into the calcified cartilage layer; and 6=disorganization (chaotic structure, clusters, and osteoclasts activity). Cellular abnormalities were scored on a scale of 0~3, where 0=normal; 1=hypercellularity, including small superficial clusters; 2=clusters; and 3=hypocellularity. The matrix staining was scored on a scale of 0~4, where 0=normal/slight reduction in staining; 1=staining reduced in the radial layer; 2=staining reduced in the interterritorial matrix; 3=staining present only in the pericellular matrix; and 4=staining absent. Joint space width was estimated by sum of the nearest distance of medial and lateral tibiofemoral joints. Histological evaluation was performed by two independent experienced researchers who were blinded to treatment arm.
Human chondrocyte isolation and culture
The samples used for this project were collected under approval of the Seoul St. Mary's Hospital (Seoul, Korea) IRB. Articular cartilage samples were obtained as surgical waste tissues from 3 patients undergoing total knee replacement surgery. The isolated chondrocytes were used for experiments within the first three passages. Cartilage was harvested from non-lesional areas, further minced, and subjected to pronase and collagenase digestion to isolate primary chondrocytes, similar to previously published Methods (16). Chondrocytes were maintained in Dulbecco's modified Eagle's medium (DMEM) medium containing 10% FBS. To activated chondrocyte conditions, the chondrocytes were stimulated with IL-1β (20 ng/ml) (R&D Systems) for 48 h. The cells were pretreated with UDCA for 2 h and then stimulated under the required activating conditions.
Real-time polymerase chain reaction (RT-PCR)
Messenger RNA (mRNA) was extracted using TRI Reagent (Molecular Research Center) according to the manufacturer's instructions. Complementary DNA was synthesized using a SuperScript Reverse Transcription system (Takara). A Light-Cycler 2.0 instrument (software version 4.0; Roche Diagnostics) was used for PCR amplification. All reactions were performed with LightCycler FastStart DNA Master SYBR Green I (Takara) according to the manufacturer's instructions. The following primers were used for human genes: for MMP3, 5'CTC ACA GAC CTG ACT CGG TT-3' (sense) and 5'-CAC GCC TGA AGG AAG AGA TG-3' (antisense); for MMP13, 5'-CTA TGG TCC AGG AGA TGA AG-3' (sense) and 5'-AGA GTC TTG CCT GTA TCC TC-3' (antisense); for ADAMTS5, 5'-TAT GAC AAG TGC GGA GTA TG-3'(sense) and 5'-TTC AGG GCT AAA TAG GCA GT-3' (antisense) and, for β-actin, 5'-GGA-CTT-CGA-GCA-AGA-GAT-GG-3' (sense) and 5'-TGT-GTT-GGG-GTA-CAG-GTC-TTT-G-3' (antisense). Messenger RNA levels were normalized to that of β-actin. An ABI Prism 7700 sequence detection system (Applied Biosystems).
MTT assay
Cell viability was assessed with the MTT assay based on the ability of mitochondria of viable cells to convert soluble MTT into an insoluble purple formazan reaction product. MTT (Sigma) solution (5 mg/ml in Dulbecco's modified Eagle's medium (DMEM) without phenol red) was added to cells in tissue culture for 2 h. The MTT solution was aspirated, dimethyl sulfoxide (DMSO) was added (200 ml/each well of 12 well plate) to solubilize formazan and detection occurred by addition of 100 ml of the reaction mixture to a 96 well plate format and reading at optical density (O.D.) 540 nm. MTT assay results for the peripheral blood mononuclear cell were derived from two independent experiments, performed in duplicates.
Statistical analysis
The change of pain behavior is expressed as means±standard error of the mean (S.E.M.). Each value of histological assessments and pain behaviors was represented as a dot plot. One-way analysis of variance followed by Bonferroni's post-hoc test was used to compare pain and histological scores. To assess the Gaussian distribution and the equality of variance, Shapiro-Wilk test and Levene's test were used, respectively. The program used for the statistical analysis was SPSS statistical software package standard version 16.0 (SPSS Inc., Chicago, IL, USA). p-values less than 0.05 (two-tailed) were considered significant.
RESULTS
The effect of ursodeoxycholic acid on pain production in OA rats
Because pain is the predominant symptom of OA, the secondary tactile allodynia in MIA-induced OA rats was assessed. The treatment of UDCA was initiated on the day of MIA injection and maintained for 2 weeks. In the von Frey hair assessment test, the paw withdrawal latency (PWL) and the paw withdrawal threshold (PWT) were prolonged significantly in the inflamed hind paw of the rats given oral UDCA (50 mg/kg of body weight) compared with the place group (Fig. 1), demonstrating the antinociceptive property of UDCA.
Microscopic findings and modified Mankin scores in joints
As the cartilage degeneration is the main histologic feature of OA joints, the chondroprotective potential of UDCA in OA joints was investigated. The isolated knee joints from the three groups were analyzed miscroscopically. In the normal joints, staining with HE, Safranin O, and toluidine blue showed a smooth surface of the articular cartilage, normal cellularity, and intensive Safranin O and toluidine blue staining. In contrast, the joints from the MIA-induced OA rats showed joint space narrowing with a marked depletion of proteoglycan. These histomorphological changes in the cartilage were reduced in UDCA-treated OA rats. The overall modified Mankin scores were significantly lower in UDCA-treated rats with OA than in vehicle-treated OA rats (Fig. 2A). Subchondral bone has been suggested to play a crucial role in the pathogenesis and progression of OA (17). Involvement of subchondral bone emerged in MIA-induced OA model where the numbers of osteoclasts were increased at by day 7 after MIA injection (18). Compared to the MIA-induced OA rats, the number of osteoclasts in subchondral bone lesion was significantly decreased by oral UDCA treatment (Fig. 2B).
Effects of UDCA on IL-1β and IL-6 expression in the cartilage of UDCA-treated OA rats
IL-1β and IL-6 have been revealed to be the main proinflammatory cytokines involved in the pathophysiology of OA (19). The results showed that the number of chondrocytes stained positive for IL-1β and IL-6 increased in the MIA-induced OA cartilage. The percentages of IL-1β-positive and IL-6-positive chondrocytes were significantly lower in the UDCA-treated group than in the vehicle-treated group (Fig. 3).
Reduced expression of nitrotyrosine and inducible nitric oxide synthase (iNOS) in subchondral bone region in OA rats treated with UDCA
To determine the degree of oxidative damage in the knee joints of UDCA-treated OA rats, immunohistochemistry was used to assess the expression of nitrotyrosine and iNOS on day 14 after MIA injection. Because subchondral bone changes develop 7 days after intra-articular injection of MIA, the changes in oxidative stress markers in the subchondral bone region was assessed. The expression of nitrotyrosine and iNOS increased markedly in the MIA-injected joints than in those from the control animals. The treatment with UDCA reduced the expressions of nitrotyrosine and iNOS in the subchondral bone (Fig. 4).
The inhibiting property of UDCA on catabolic factors in human OA chondrocytes
We next investigated the possible role of UDCA in modulating the expression of catabolic molecules in human OA chondrocytes. The results show that the gene expression of MMP-3, -13, and ADAMTS-5 increased after the addition of IL-1β and that the effect was reduced after the treatment with UDCA (Fig. 5A). MTT assay was used to determine whether or not chondrocytes viability is affected by UDCA treatment. UDCA treatment up to 50µM in human OA chondrocytes did not affect cell viability (Fig. 5B).
DISCUSSION
This study demonstrated that oral administration of UDCA attenuated the pain severity and cartilage loss, and reduced oxidative stress in subchondral bone legion in an animal model of OA. The chondroprotective property of UDCA may be achieved by reduced expression of proinflammatory molecules and attenuated catabolic activities of chondrocytes.
Until now, the main target of OA treatment is to control chronic pain. Although cartilage degeneration is the main histological feature of OA, the extent of cartilage loss has not been positively related with pain severity in the patients with OA. MIA-induced OA rat model used in the present study mimics pain and biochemical/structural changes associated with human OA (20). Recent studies have suggested that proinflammatory cytokines such as IL-1β are implicated in pain generation in OA (21). Recent clinical studies using magnetic resonance imaging suggested that pain severity of human OA is positively correlated with the changes of subchondral bone (22,23). Interestingly, animal studies showed that nitric oxide donor significantly increased subchondral bone sclerosis during subchondral remodeling suggesting that oxidative stress could contribute to pain generation in OA. Our study demonstrating that UDCA treatment could significantly attenuated the extent of oxidative damage in subchondral bone may suggest that oxygen-radical scavenging material could be a therapeutic candidate to control pain generation in OA.
The pathophysiology of OA development and progression is complex and poorly understood. The current paradigm of OA has changed to include an active biological process of matrix degradation leading to the release of MMPs and eventually cartilage loss; both acute and chronic inflammatory processes propagate disease process (19). MMPs have been shown to degrade all components of the extracellular matrix and many studies have reported that increased activities of interstitial collagenases MMP-3 and MMP-13 in human OA cartilage as well as experimental animal models of OA (24-26). As one of catabolic factors in articular cartilage, there is ADAMTS5 that is one of aggrecanases. In conjunction with MMPs, ADAMTS5 can function to degrade extracellular matrix components of cartilage. Interestingly, ADAMTS5 is superior to MMPs in the aspect of cleaving aggrecan core protein in articular cartilage (27). Song et al. recently demonstrated that suppression of ADAMTS5 in human chondrocytes can attenuate the degradation of aggrecan in cartilage (28). In our study, the expression of MMP-3, MMP-13 and ADAMTS5 was significantly inhibited by UDCA in the IL-1β-stimulated human chondrocytes, suggesting the potential of UDCA as one of the DMOADs in OA treatment.
Continuous and low-grade oxidative stress to cells and matrix contribute to OA pathogenesis. Increased oxidative stress associated with the aging process creates chondrocytes to become more susceptible to oxidant-mediated cell death through the dysregulation of the glutathione antioxidant system (29). Therefore, reducing oxidative stress could be a novel therapeutic strategy to inhibit cartilage degeneration and to relieve debilitating pain in OA patients. Long-term treatment with UDCA markedly improved biochemical and histopathological indices of liver damage and prolonged survival (30,31). In addition to the clinical benefits of UDCA, the immunological mechanisms underlying UDCA efficacy remained uncertain. Interestingly, Okada et al. reported that UDCA can induced nuclear factor E2-related factor 2 (Nrf2) expression level in hepatocytes suggesting its potential to stimulate antioxidative stress systems (32). Nrf2 is a key transcription factor that plays a central role in the protection of cells against oxidative stresses. Nuclear translocation of Nrf2 up-regulates several genes encoding antioxidant proteins including heme oxygenase-1 (HO-1) and NAD(P)H (33). Taken together, UDCA may exert the anti-nociceptive and chondroprotective properties in OA animal models through the down-regulation of oxidative damage in subchondral bone legion.
In conclusion, this is the fires study demonstrating the preventive potential of UDCA in the MIA-induced OA model. UDCA reduced pain and ameliorated cartilage destruction via reducing oxidative stress and inhibiting catabolic factors implicated in disturbed extracellular matrix homeostasis of OA cartilage. Considering its antioxidant and anti-inflammatory property of UDCA, this may be a promising therapeutic option for OA.
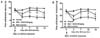




 XML Download
XML Download