

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Asthma is a chronic lung disease afflicting over 300 million people and its prevalence is increasing (1). Airway inflammation is a major symptom of allergic asthma and can lead to the remodeling of airway walls, resulting in airway obstruction, mucus hypersecretion and airway hyperreactivity (2,3). It is generally accepted that Th2-immune responses to allergen are responsible for the development of allergic asthma (4). Th2-mediated immune responses favor humoral antibody production, characterized by the secretion of IL-4, IL-5, and IL-13, and these cytokines are essential for the development of allergen-specific IgE producing-B cells (4). Antigen-specific IgE interacts with mast cells and basophils via the high-affinity Fcε receptor (FcεRI), resulting in the release of histamine, prostaglandin, leukotrienes, proteases, growth factors, cytokines, and chemokines that exacerbate asthma (5).
Many studies have attempted to elucidate the relationship between allergic asthma and infectious disease. Epidemiologic studies have reported a considerably lower prevalence of allergic disease in developing countries; in addition, incidence of childhood infections shows consistent negative association with atopy and allergic disease (6,7). Exposure to pathogens, such as hepatitis A virus, Toxoplasma gondii, and Helicobacter pylori reduces the risk of atopy by >60% (8). The so-called "hygiene hypothesis" has been proposed to explain these observation. Insufficient stimulation of Th1 cells induced by limited exposure to bacterial and viral pathogens during early childhood cannot counterbalance the expansion of Th2 cells and results in a predisposition to allergy (6). These studies suggested the possibility that immune response induced by infection is related to allergic asthma. Hence, more research regarding the effect of immune response induced by microbial components on allergic airway inflammation is required to develop novel therapeutic strategies for asthma.
Toll-like receptors (TLRs) express on various cell types and associated with recognition of microbial components known as pathogen-associated molecular patterns (PAMPs) (9). TLRs are comprised of extracellular leucine-rich repeats responsible for the ligation and cytoplasmic Toll/interleukin-1 receptor (TIR) domains required for initiating intracellular signaling (10). The ligation of TLRs initiates immune responses by activating downstream transcription factors, including NF-κB, MAPK, and IRFs, which lead to the production of inflammatory cytokines such as TNF-α, IL-1β, and IL-6 (11). Signal transduction pathways of TLRs are further classified into the myeloid differentiation primary response 88 (MyD88)-dependent pathway and the MyD88-independent pathway. TIR-domain-containing adapter-inducing interferon-β (TRIF) is required for MyD88-independent signal transduction of TLR3 and TLR4 (12,13).
Respiratory infections may either prevent or facilitate asthma, thereby implicating a role for TLRs signaling in the regulation of Th2-driven airway disease (14). Many studies have investigated the role of TLR3 and TLR4 in allergic asthma. TLR3 activation by virus-derived dsDNA results in Th2 cytokine production and influx of eosinophils, myeloid DCs, and inflammatory T cells, and stimulation with poly (I:C) increases in the total number of cells in bronchoalveolar lavage (BAL) fluid (15,16). Stimulation of TLR4 expressed by airway stromal cells promotes cytokine production that mediates the maturation of Th2-polarized lung DC (17). Th2-mediated immune response and the asthma phenotype may also be enhanced by exposure to LPS and activation of the TLR4 pathway in a MyD88-dependent manner (18,19).
A role for MyD88-independent TRIF signaling in the development of Th2 responses and allergic airway inflammation has been suggested by studies on viral myocarditis and LPS stimulation of bone marrow derived dendritic cells (20,21). However, the role of TRIF in allergic airway inflammation is not yet fully understood. In this study, we determined the effect of TRIF deficiency on allergic airway inflammation by a mouse model of OVA-induced allergic airway inflammation.
MATERIALS AND METHODS
Animals and reagents
C57BL/6 mice were obtained from KOATECH (Pyeongtaek, Gyeongi-do, Korea). TRIF-/- mice with C57BL/6 background were kindly gifted by Shizuo Akira (Osaka University, Japan). Albumin from chicken egg white (OVA) and aluminum hydroxide were purchased from Sigma-Aldrich (St. Louis, MO, USA). All experiments were approved and carried out under the supervision of the Institutional Animal Care and Use Committee at Konyang University.
Animal sensitization and treatments
Protocols for inducing allergic airway inflammation in mice are depicted schematically in Fig. 1. Briefly, wild-type (WT) mice and TRIF-/- mice were sensitized by administering 40 µg OVA with 4 mg aluminum hydroxide intraperitoneally (i.p.) in a volume of 200 µl on days 0 and 7. Anesthetized mice were challenged intranasally (i.n.) with 200 µg OVA diluted in PBS in a volume of 25 µl. Control groups received PBS alone. Animals were sacrificed 2 days after the final i.n. administration, and samples of lung, serum, and BAL fluid were collected for further analysis.
Measurement of the concentration of cytokines and OVA-specific serum antibodies
Concentrations of IL-5 and IL-13 in lung extract were measured using a commercial enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Abingdon, UK). OVA-specific serum IgE, IgG1, and IgG2c levels were determined by ELISA. OVA (10 µg/ml) was coated onto 96-multiwell plates. Serum samples were diluted 1/10,000 for IgG1, 1/1,000 for IgG2c and 1/20 for IgE. Biotinylated rat anti-mouse IgE (BD Biosciences, San Jose, CA, USA) was applied, followed by Streptavidin-HRP (BD Biosciences) to quantify OVA-specific serum IgE. Peroxidase-conjugated rat anti-mouse IgG1 and IgG2c (Southern-Biotech, Birmingham, AL, USA) were used to quantify OVA-specific serum IgG1 and IgG2c respectively.
Quantification of cells in BAL fluids
BAL fluid was prepared by washing the lungs with 0.8 ml of PBS. The cell pellets were prepared by centrifugation at 300 ×g for 3 min. After discarding the supernatants, cell pellets were resuspended in RPMI 1640. Cells were stained with trypan blue, and the total number of viable cells was determined using a hemocytometer.
Histological evaluation of tissue inflammation
To evaluate tissue inflammation, the left lung from each mouse was fixed in 10% neutral-buffered formalin (pH 7.0) for 48 hours and embedded in paraffin. Sections (2-µm thick) were prepared and stained with hematoxylin and eosin. Tissue inflammation in each sample was examined by light microscopy and was expressed as a numerical score. Tissue inflammation based on the abundance of lesions was scored as follows: 0=non-specific lesion, 1=slight, 2=mild, 3=moderate, 4=marked, and 5=severe.
Statistical analysis
Differences among the mean values of the different groups were analyzed, and the values were expressed as mean±SD. Statistical analyses were performed by Student's t-test by GraphPad Prism version 4 (GraphPad Software, San Diego, CA, USA). Values of p<0.05 were considered significant.
RESULTS
TRIF deficiency does not contribute to histopathological changes in an OVA-induced model of airway inflammation
Inflammation in tissues was assessed by histological examination, and the total number of cells in BAL fluid was counted (Fig. 2). OVA-sensitized/challenged WT and TRIF-/- mice showed inflammatory cell infiltration around the airways and interstitium of the alveoli (Fig. 2A). These lesions did not appear in PBS-sensitized/challenged control WT and TRIF-/- mice. Scores indicating severity of inflammation were significantly increased in OVA-sensitized/challenged WT and TRIF-/- mice compared to PBS-sensitized/challenged WT and TRIF-/- mice; however, a significant difference in the scores was not observed between OVA-sensitized/challenged WT mice and OVA-sensitized/challenged TRIF-/- mice (Fig. 2B). The total number of cells in BAL fluid was also increased in OVA-sensitized/challenged WT and TRIF-/- mice, and no significant difference in the total number of cells was detected between the two groups (Fig. 2C).
TRIF deficiency does not change the level of inflammatory cytokines in OVA-induced allergic airway inflammation
Th2 cytokines are associated with the pathogenesis of allergic response, whereas Th1 cytokines act to counter-balance of effect of Th2 cytokines (6). IL-5 is the principal eosinophil-activating cytokine and also mediates eosinophil recruitment and triggers the activation of B cells (15,22,23). IL-13 plays a role in the contraction of smooth muscle cells in the airway (24). IL-12 prevents the expansion of Th2 cells, and IFN-γ is the major cytokine produced by Th1 cells (25,26). To evaluate cytokine production in lung tissue, the concentration of cytokines in the supernatant of lung homogenate were measured by ELISA. The level of IL-5 and IL-13 in lung extract were increased in both WT and TRIF-/- OVA-sensitized/challenged WT and TRIF-deficient mice (Fig. 3A and B). The level of type 1 cytokine IL-12 was also increased in both WT and TRIF-/- OVA-sensitized/challenged mice, and no significant difference was observed between the groups (Fig. 3C). Finally, IFN-γ was not significantly increased in OVA-sensitized/challenged mice compared to control mice (Fig. 3D).
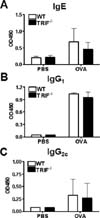
TRIF deficiency does not affect to serum levels of OVA-specific IgE, IgG1, and IgG2c in the OVA-induced model of airway inflammation
Serum levels of OVA-specific IgE, IgG1 and IgG2c were measured by ELISA. Based on our previous results, we expected that TRIF deficiency would not affect the level of allergen-specific immunoglobulin. OVA-specific IgE, IgG1, and IgG2c levels in OVA-sensitized/challenged TRIF-/- mice were similar to those found in WT mice (Fig. 4).
DISCUSSION
Infections are closely related to the pathogenesis of allergic asthma (27). However, the effect of infections on the development of allergic asthma is quite controversial. Viral infection of the airway commonly aggravates allergic asthma (27). Human rhinovirus (HRV) is associated with the exacerbation of asthma in both children and adults (28,29). In addition, respiratory syncytial virus (RSV) infection induces a lower IFN-γ/IL-10 ratio and contributes to the polarization of Th2-biased immune responses and the production of IL-13-mediated AHR (27,30). The role of bacterial infection in allergic asthma is controversial. Recent studies showed that bacterial infections are associated with the exacerbation of asthma and exposure to LPS enhances the Th2 response to inhaled allergens (14,31,32,33,34,35). In contrast, epidemiological studies suggest that insufficient Th1 immune response induced by infection in infancy is related to development of allergic disease (6). In addition, exposure to airborne endotoxin attenuated asthma by promoting enhanced Th1 response and tolerance to allergens (36).
TLR signaling has been suggested as a basal mechanism that connects the infection and allergic disease and emerged as a novel therapeutic target of drugs for asthma (14,15,16,17,18,19,37). To develop novel drugs for asthma, the role of molecules associated with TLR signaling in allergic asthma must be defined. Although the role of TRIF, an adaptor molecule of TLR3 and TLR4, which recognize viral RNA and bacterial LPS, respectively, in allergic asthma has been studied (15,38,39), its function is not yet fully understood. Deficiency of TRIF shows contrasting effects, depending on the animal model and experimental conditions used. In a pollen-induced asthma model, TRIF deficiency results in the exacerbation of airway inflammation by augmenting the total number of BAL inflammatory cells and increasing of chemokines KC and eotaxin in BAL fluid compared with those in BAL fluid of WT mice (38). However, TRIF deficiency reduces IL-17 associated with neutrophilic asthma in an OVA-induced model of allergic airway inflammation (40). Interestingly, in some studies, TRIF deficiency did not have any effect on airway inflammation or the asthmatic phenotype in murine models of OVA-induced asthma (15,18).
In our study allergic airway inflammation induced by immunization with an i.p. injection of OVA/alum and subsequent challenge with an i.n. injection of OVA solution was developed to determine the effect of TRIF deficiency on allergic airway inflammation. The histopathological scores, total inflammatory cells in BAL fluid, the production of Th1 and Th2 cytokines in lung tissue, and levels of OVA-specific immunoglobulin in TRIF-/- mice were comparable to those observed in WT mice. These results suggest that TRIF deficiency did not affect on the development of airway inflammation in OVA in murine models of OVA-induced asthma.



 XML Download
XML Download