

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abbreviations
ILCs
innate lymphoid cells
AHR
airway hyperreactivity
BAL
broncho alveolar lavage
NCR
natural cytotoxicity receptor
Lti
Lymphoid Tissue inducer
RORα
retinoic acid receptor-related orphan receptor α
RORγt
retinoic acid receptor-related orphan gamma t
ID2
inhibitor of DNA binding 2
OVA
ovalbumin
HDM
house dust mite
RSV
respiratory syncytial virus
TSLP
Thymic stromal lymphopoietin
INTRODUCTION
Traditionally, most scientists believed that asthma was an immunological disease mainly mediated by Th2 cells and the adaptive immune system (1). Indeed, Th2 cells do play a critical role in allergic asthma, mostly through their production of an array of cytokines: IL-4, a switch factor for IgE; IL-5, a growth and differentiation factor for eosinophils; and IL-13, a direct causative agent of airway hyperreactivity (AHR) that causes contraction of airway epithelial cells and airway smooth muscle cells. However, over the past few years, it has become clear that asthma is actually a heterogeneous and complex disease, and other immune cells as well as Th2 cells can play major roles in inducing asthma (2). For example, therapeutic trials targeting Th2 cytokines benefitted only a portion of asthma patients, demonstrating the involvement of other immune cells in asthma (3).
The most commonly studied form of asthma is allergic asthma, which is triggered by continuous exposure to allergens. As mentioned above, Th2 cells and eosinophils are at the center of this pathogenesis. The other type of asthma, non-allergic asthma, is mainly associated with exposure to environmental factors such as air pollution, ozone, cigarette smoke, diesel particles, viral infection, stress, and nutritional diseases such as obesity (2). Neutrophils and other innate immune cells in the airway seem to play key roles in inducing non-allergic asthma (4,5,6,7). Genetic factors also seem to influence the development of intrinsic forms of asthma. This heterogeneity suggests that in addition to the allergic and non-allergic forms of asthma, there may be even other forms of asthma mediated by immune cells that regulate and shape pulmonary inflammation (2). For example, recently identified innate lymphoid cells (ILCs) were found to regulate asthma through pathways that do not involve Th2 cells. In this review, we focus on the role of ILCs in inducing several forms of asthma as well as recent findings regarding these newly described immune cells.
INNATE LYMPHOID CELLS
ILCs are non-T, non-B effector cells that show high levels of effector cell functions (mainly cytokine production) upon activation. ILCs were first identified in the fat associated lymphoid cluster (FALC) and the intestinal tract (8,9,10), and shortly thereafter they were also identified in the lung (11,12,13). These cells were shown to be involved in tissue homeostasis, repair, and remodeling as well as innate immunity (14). As non-T, non-B effector cells, ILCs lack rearranged antigen-specific receptors (T cell and B cell receptors) and are therefore antigen nonspecific, but they react rapidly to a wide range of innate signals. Given that ILCs produce a great variety and amount of cytokines, it is surprising that they were unidentified earlier, but this might have been due to the focus of immunologists on adaptive immune cells or to technical limits on the ability to identify different cell populations.
ILCs have been classified into three subsets-type 1(ILC1s), type 2(ILC2s), and type 3(ILC3s)-based on their cytokine production (Table I) (15). The prototypic ILC1s is the NK cell, which produces IFN-γ and expresses T-bet (T-box transcription factor) (16,17). ILC2s (previously called natural helper cells or nuocytes) were discovered more recently; these cells produce IL-5, IL-9, and IL-13, a set of cytokines similar to those produced by Th2 cells (8,9). ILC2s require the transcription factors RORα (retinoic acid receptor-related orphan receptor α) (18,19) and GATA3 (20). Among type 3 ILCs (ILC3s), there are at least three different subtypes: (a) lymphoid tissue inducer (LTi) cells, which produce both IL-17 and IL-22 (21); (b) IL-17-producing cells, which are active in the gut of patients with colitis (22) and in obesity-related forms of asthma (23); and (c) IL-22-producing cells, which are present in the skin (24,25) and gut (26).
Although there are different subsets of ILCs, all share a common lymphoid progenitor, which has been identified as Lin-IL-7Rα+Kit+/lowSca-1+/low, and they also share common developmental pathways (14,19,27). All ILCs require IL-7 signaling and Id2, a transcriptional repressor which regulates Notch signaling, for proper development (28,29,30,31). These findings indicate that ILCs might develop from a common progenitor and then differentiate into different subsets based on the situation. However, several studies have also suggested that additional pathways and plasticity might exist in ILC lineage establishment (31).
ROLES OF ILC1s IN THE DEVELOPMENT OF ASTHMA
ILC1s
ILC1s express the transcription factor T-bet and produce IFN-γ, a type 1 cytokine. Through their production of IFN-γ, ILC1s show antimicrobial activity and are involved in the clearance of intracellular infections. Compared to ILC2s and Ilc3s, ILC1s have not been well defined. They currently include IL-7Rα- NK cells, because NK cells also express T-bet and produce IFN-γ; however, it is not clear whether a non-NK, IL-7Rα+ ILC1 lineage exists. A recent study identified a distinct ILC1 lineage in the intestinal lamina propria that expresses NKp46, NK1.1, and T-bet but lacks Eomesodermin, RORγt, and fate map. Interestingly, this newly described subset of ILC1s is the main producer of IFN-γ and TNF-α in response to IL-12 and intestinal infection by Toxoplasma gondii (32). Therefore, these results suggest that there might be an additional ILC1 subset distinct from NK cells.
ILC1s in asthma
Although the ILC1 lineages have not been well characterized, NK cells belong to the ILC1 family and have been studied thoroughly in the past. Therefore, we will focus on NK cells in the following discussion as analogous to the role of ILC1s in the development of asthma. In the context of asthma, NK cells have not been studied extensively and their roles are still not clear. This might be due to limitations of the methods used for specific depletion of NK cells. However, recent studies have shown that NK cells play an important role in the development of several forms of asthma. NK cells are responsible for lung inflammation during viral infections (33,34) and may regulate the pathogenesis of virus-induced asthma. For example, when humans and mice are infected with rhinoviruses, which cause a range of respiratory diseases, significantly higher amounts of IL-15 and IL-15Rα are detected in fluid from the nasal mucosa and in bronchial tissues (35). Since IL-15 is essential for the activation of NK cells as well as the development of ILC1s, it is possible that ILC1s are involved in the development of virus-associated asthma.
The role of NK cells in allergic asthma has been better studied. Korsgren et al. showed that depletion of NK cells using anti-NK1.1 antibody reduced asthma symptoms. In mice depleted of NK cells, eosinophilic infiltration and allergen-specific IgE production in the lung were greatly reduced (36). However, depletion of NK cells using anti-NK1.1 antibody can also deplete natural killer T cells, which are essential for the development of AHR. Therefore, it was not clear whether NK cells themselves could regulate the development of asthma. However, more recent studies of ovalbumin (OVA)-induced AHR have shown that NK cells are more numerous in OVA-challenged airways. Moreover, perforin-deficient mice failed to develop OVA-induced AHR, suggesting that NK cells are essential for priming OVA-induced AHR (37). Another study was done with a house dust mite (HDM)-induced asthma model. Allergic airway inflammation was induced by intranasal administration of HDM extract in wild-type mice and in NKG2D-deficient mice, which lack the receptor that activates NK cells. NKG2D-deficient mice were resistant to the development of HDM-induced AHR and showed reduced levels of eosinophils, serum IgE, and Th2 cells. Furthermore, transfer of NK cells into NKG2D-deficient mice restored HDM-induced airway inflammation, suggesting that NK cells might be critical for the development of allergen-induced AHR (38). Taken together, these studies suggest potential roles of ILC1s in the pathogenesis of asthma.
ROLES OF ILC2s IN THE DEVELOPMENT OF ASTHMA
ILC2s
ILC2s were first reported among ILC lineages as non-T, non-B cells that proliferated upon administration of IL-25 (39,40) and induced Th2-like immune responses such as increased serum IgE level and eosinophil infiltration in the digestive tract. Non-T, non-B lymphocytes producing IL-5 and IL-13 were also detected in the lungs of patients with asthma, but their specific functions were unclear at the time (41). Then, in 2010, three different groups characterized ILCs that produced large quantities of IL-5 and IL-13 in response to IL-25 and IL-33 (8,9,42). The initial report indicated that these cells were found in lymphoid compartments of adipose tissue and in gut-associated lymphoid tissue. Although these cells had different surface markers and functions, they also shared some common surface markers and cytokine secretions (43). Now, a consensus has been reached to refer to all of these cells as ILC2s (8,9,40,44).
The cytokines IL-7, IL-25, and IL-33 seem to be important for the development of ILC2s. Transcription factors such as Id2, RORα, and GATA3 as well as Notch signaling also contribute to the development of ILC2s (18,19,27). Although there were subtle differences between the cells described in the previous reports, it seems that mouse ILC2s generally express CD25, CD90, and variable amounts of CD117 and CD127 and play critical roles in type 2 immune responses. In addition, ILC2s express variable amounts of CD278, ST2 (IL-1RL1), and IL-17BR.
ILC2s in virus-induced asthma
Th2 cells are believed to be at the center of allergic asthma pathology. Thus, previous studies of asthma paid little attention to other cell types. However, there is evidence that other cell types are important for inducing asthma. In human patients with asthma, viral infections can precipitate virtually all asthma symptoms regardless of the presence of allergy, suggesting that innate cells rather than adaptive cells (such as Th2 cells) are critical for this type of asthma. For example, Il1rl1-/- mice, which lack the IL-33 receptor, developed near-normal AHR responses after sensitization and challenge with OVA. However, when Il1rl1-/- mice were infected with influenza A virus (H1N1), AHR was greatly reduced (11). Even in the absence of adaptive immunity (e.g., in Rag2-/- mice), AHR was induced by influenza A virus. This airway hyperreactivity depended on ILC2s expressing c-Kit, Sca-1, Thy1.2 (CD90), and ST2 (11). Moreover, depletion of ILC2s in Rag2-/- mice using anti-Thy1.2 antibody abolished the influenza-induced AHR response. These responses were dependent on IL-13 production, since adoptive transfer of IL-13-secreting ILC2s restored influenza-induced AHR in Il13-/- recipients. In another study, Hong et al. infected 6-day-old neonatal mice with rhinovirus and found that the virus promoted IL-25 production, which in turn induced activation of ILC2s. In addition, proliferation of IL-13-secreting ILC2s was observed in rhinovirus-infected neonatal mice but not in adult mice. Treatment with anti-IL-25 neutralizing antibody attenuated ILC2 proliferation, mucous hypersecretion, and airway responsiveness (45). It has also been suggested that infection with respiratory syncytial virus might result in the subsequent development of asthma, independent of allergen challenge (46).
The studies described above show that ILC2s play a pathological role during virus-induced asthma. However, another study has suggested that ILC2s may also contribute to lung homeostasis. In mice infected with influenza virus (PR8), ILCs accumulated in the lung, and depletion of ILCs resulted in loss of airway epithelial integrity, diminished lung function, and impaired airway remodeling. ILCs seem to contribute to lung homeostasis by producing amphiregulin (47). Taken together, these results indicate that ILC2s may have a dual function, either inducing inflammation or restoring airway cell integrity depending on the strength of the pathogen and the length of infection.
ILC2s in allergic airway disease
As mentioned previously, ILC2s secrete high levels of IL-5 and IL-13, which suggests they might play an important role in allergic asthma as well as virus-induced asthma, since type 2 cytokines are believed to play a critical role in the development of allergen-induced AHR. In contrast to the virus model, Il1rl1-/- mice had normal AHR responses after sensitization and challenge with OVA, suggesting that ILC2s may not be required for allergen-induced AHR, although ILC2s clearly accumulated in the lungs during allergen challenge (11). Using an OVA sensitization and airway challenge method, Kearly et al. showed that persistent AHR was correlated with the continued presence of Th2 cells (48). They found that the ST2-IL-33 pathway was important, since antibody against ST2 reduced IL-4 and IL-13 production as well as allergic inflammation and AHR. They concluded that Th2 cells are the primary cell type that cause AHR. However, ILC2s also rely on the ST2-IL-33 pathway, therefore, they are capable of inducing type 2 allergic airway inflammation.
While studies with allergen-induced asthma models have focused on allergen-specific Th2 cells, ILC2s also contribute to type 2 allergic airway disease. Treatment with recombinant IL-25 or IL-33 induced the proliferation of IL-5- and IL-13-producing ILC2s in the lungs and mediastinal lymph nodes (49,50,51,52,53). Klein et al. showed that both ILC2s and Th2 cells are the major sources of IL-5 and IL-13 in HDM-induced and OVA-induced asthma (53). Halim et al. showed that intranasal administration of protease-containing allergens (papain) induced eosinophilia and mucus hyperproduction in Rag1-/- mice (which have ILCs) but not Rag2-/- Il2rg-/- mice (which lack all types of ILCs). Adoptive transfer of ILC2s enabled Rag2-/- Il2rg-/- mice to respond to papain. Papain damages stromal cells, which can then release IL-33 and thymic stromal lymphopoietin (TSLP), which in turn can activate ILC2s (54). Wilhelm et al. administered papain into the lungs and found IL-9-producing ILC2s, a new subset of ILC2s (55). These IL-9-producing cells required IL-2 and IL-33 (but not IL-25) to produce IL-5 and IL-13, and blockade of IL-9 resulted in reduced expression of IL-5 and IL-13, suggesting that ILC2s may initially produce IL-9 and then mature into IL-5- and IL-13-producing cells. In a papain-induced asthma model, ILC2s were the main source of type 2 cytokine production.
Another ubiquitous fungal allergen, Alternaria alternata, also induced IL-33 production in the lung, which in turn induced the proliferation of Lin-CD25+CD44hi ILC2s. The production of IL-5 and IL-13 by ILC2s in response to A. alternata was nearly abolished in Il1rl1-/- mice, suggesting that ILC2s mediate asthma in an IL-33-dependent manner (50). While all of these studies together suggest that ILC2s play essential roles in allergen-induced airway disease, at this point it is unclear whether ILC2s alone are sufficient for inducing allergic asthma. Further study of the precise requirement for ILC2s in allergic lung disease is needed.
ROLES OF ILC3s IN THE DEVELOPMENT OF ASTHMA
ILC3s
RORγt is a ligand-dependent nuclear hormone receptor expressed in several cell types such as Th17 cells and the type 3 ILCs (ILC3s) (29,56,57). RORγt-dependent ILCs can be divided into at least three different subpopulations based on their functional characteristics. LTi cells, the first subpopulation to be identified among the type 3 ILCs, represent the prototypic cell type of the RORγt+ family of ILCs (58). These cells are RORγt+ and IL-7Rα+ and are mainly involved in secondary lymphoid organ formation (59). Approximately half of fetal LTi cells express CD4, while half of them do not, suggesting that there might be a precursor-progeny relationship between CD4- and CD4+ LTi cells (60). The second group of ILC3s is the IL-17-producing ILC3s. These cells were found in the intestine of fetal mouse (61) and in human fetal lymph nodes (62). IL-17-producing ILC3s are also present in the intestine of adult mice (10) and humans (63) under inflammatory conditions. IL-17-producing ILC3s do not express NKp46, suggesting that these cells are distinct from IL-22-producing NKp46+ cells. The third group of ILC3s is the IL-22-producing ILC3s. IL-22-producing ILC3s express the natural cytotoxicity receptor (NCR), which is NKp44 in humans (64) and NKp46 in mice (65,66). Owing to their NK-associated receptor expression, the ILC3 and ILC1 (NK cells) lineages are considered closer to each other than to the ILC2 lineage; however, expression of RORγt and production of type 3 cytokines such as IL-17 and/or IL-22 should be considered standard markers of type 3 ILCs.
ILC3s in obesity-associated asthma
Asthma is one of the representative immune diseases induced by type 2 cytokines produced by Th2 cells and ILC2s. The role of IL-17A or IL-22 in the development of asthma is controversial, since IL-17A may either inhibit or exacerbate allergic asthma (5,67). However, recent studies have indicated that IL-17A can directly cause AHR (68,69). These studies have shown that cytokine production by Th17 cells or direct administration of recombinant IL-17 can induce airway inflammation and AHR by inducing contraction of smooth muscle cells. Therefore, IL-17 might have a pathogenic role in airway disease, particularly non-allergic asthma.
Obesity is usually associated with several diseases, such as type 2 diabetes mellitus, cardiovascular disease, liver disease, and cancer (70). Interestingly, obesity is also a major risk factor for the development of asthma, particularly a severe, steroid-resistant form of asthma (71,72). Thus, these findings point to unknown mechanisms of non-allergic asthma that differ from those of allergic asthma.
Interestingly, some obese people show asthma symptoms with increasing levels of IL-17A. Recently, a role for IL-17A-producing ILC3s in AHR associated with obesity was proposed (23). Kim et al. demonstrated that obese mice fed a high-fat diet spontaneously developed AHR and had significant numbers of IL-17-producing cells in their lungs. The majority of IL-17A-producing cells were Lin-Thy1.2+Sca-1+CCR6+ILC3s. In this study, it was first shown that ILC3s were present in the lung and mediated the development of AHR. These IL-17-producing ILC3s were distinct from ILC1s and ILC2s, since they did not express T-bet or produce IFN-γ and did not produce IL-5 or IL-13. Obesity-induced AHR is independent of adaptive immune cells, since Rag1-/- mice fed a high-fat diet became obese, had more IL-17-producing ILC3s in the lungs, and developed AHR. Just as IL-33 and IL-25 are key cytokines that can induce proliferation of ILC2s, IL-1β could promote proliferation of IL-17-producing ILC3s in the lung (but not IL-22-producing ILC3s). Furthermore, adoptive transfer of IL-17-producing ILC3s into Rag2-/- Il2rg-/- mice (which lack all types of ILCs) restored IL-1β-induced AHR, indicating that IL-17-producing ILC3s by themselves can induce AHR. The production of IL-1β required activation of the NLRP3 inflammasome, since Nlrp3-/- mice fed the high-fat diet became obese but did not develop AHR. Secretion of the active form of IL-1β, which is mediated by the NLRP3 inflammasome, was increased in M1 macrophages and the lungs of obese mice and induced the proliferation of IL-17-producing ILC3s. Moreover, blocking IL-1β signaling with a short treatment with anakinra (an IL-1R antagonist) abrogated development of AHR in the obese mice and greatly reduced the number of IL-17-producing ILC3s in the lung.
Although there is only one study so far showing that ILC3s are related to the development of asthma, it is possible that ILC3s may also be involved in the development of other forms of asthma, such as those induced by viruses or allergens, since Th17 and Th22 cells are considered pathogenic in asthma. However, the precise roles of each ILC lineage (ILC1s, ILC2s, and ILC3s) remain to be determined.
ILCs IN HUMANS
Although most studies regarding ILCs have been done with mice, multiple studies in human subjects suggest that ILCs are also important for the human immune system as well, especially under inflammatory conditions. Here we will summarize the studies that have been done with ILC1s, ILC2s, and ILC3s in the context of human diseases.
ILC1s in humans were first identified in the gut (73,74). These cells expressed the transcription factor T-bet and responded to IL-12. ILC1s are now characterized in humans and mice, and immunologists believe that these cells are distinct from NK cells because they do not produce perforin or granzyme B and do not express NK cell markers such as CD56, CD16, and CD94 (74). Under the influence of IL-12 (and IL-15), a subset of human ILC1s produce IFN-γ. The frequency of this ILC1 subset was greatly increased in inflamed intestine of patients with Crohn's disease (73,74), which indicated a role for IFN-γ-producing ILC1s in the pathogenesis of gut inflammation as well as inflammation of other mucosal sites.
ILC2s are the best-studied subset of ILCs in humans. An ILC2-like cell population was first reported by Allakhverdi et al. in 2009, although ILCs had not been identified at that time. They found non-T, non-B cells producing IL-13 and IL-5 in the sputum of asthmatic subjects but not in normal subjects. They also found that the non-T, non-B CD34+ cells expressed receptors for TSLP and IL-33 and responded to these cytokines very rapidly (which is characteristic of ILC2s). In addition, the number of these cells increased in response to a specific allergen inhalation challenge (41). Although Allakhverdi et al. did not further characterize this subset, their findings suggest that ILC2s may play an important role in human asthma. The source of the IL-33 and TSLP was not defined by this group, although airway epithelial cells and airway smooth muscle cells may be the key sources of IL-33 and TSLP (75).
Recent studies have also suggested that human ILC2s are important sources of IL-13 and IL-5. These studies identified ILC2s as Lin-, IL-7Rα+ cells expressing CRTH2 and CD161 (76). ILC2s also seem to be involved in the pathogenesis of several other diseases. For example, ILC2s may important roles in eosinophilic airway inflammation resulting in pleural pathology. Primary spontaneous pneumothorax, the spontaneous presence of air in the pleural space, is one of the most common causes of eosinophilic pleural effusion. Significantly higher concentrations of IL-5 and eotaxin-3 were detected in the pleural fluid of patients with primary spontaneous pneumothorax, and this was associated with the presence of IL-33 and TSLP (77). ILC2s have also been detected in the skin of patients with atopic dermatitis; these were identified as Lin-IL-7Rα+ST2+ cells, which are dependent on IL-33 or IL-25. Signaling via IL-33 induced type 2 cytokine production and amphiregulin expression by ILC2s and also induced migration of ILC2s (78). Lastly, ILC2s have been found in nasal polyp tissue from patients with chronic rhinosinusitis (79). Since TSLP is increased in nasal polyps of these patients (79) and IL-33 production and ST2 expression are increased in patients with allergic rhinitis (80), ILC2s may play an important role in human allergic rhinitis. Taken together, these human reports certainly suggest that the characteristics and functions of human ILC2s are similar to those of murine ILC2s and that these cells play a critical role in the human respiratory tract and skin.
ILC3s have also been investigated in humans, especially in patients with psoriasis. Teumissen et al. found CD117+NCR+ ILC3s in healthy peripheral blood and cultured dermal explants. NCR+ ILC3s produced IL-22 after cytokine stimulation. Remarkably, IL-1β plus IL-23 converted dermal NCR-ILC3s to NCR+ ILC3s in ex vivo culture. IL-22 is a key cytokine involved in epidermal thickening, suggesting that IL-22-producing NCR+ ILC3s may participate in the pathology of psoriasis (25). Another study tested tissue samples from non-lesional or lesional psoriatic skin and from nickel- and petrolatum-exposed skin of patients with contact allergy to nickel. They found that RORγt+CD56+ ILC3s, which are known to produce IL-22, were elevated in both non-lesional and lesional skin from patients with psoriasis as well as skin from patients with contact allergy to nickel in comparison with healthy skin. These results suggest that IL-22-producing ILC3s contribute to the pathogenesis of psoriasis in humans (81).
Additionally, examination of bronchoalveolar lavage fluid from a small group of patients with severe asthma revealed the presence of IL-17-producing ILC3s, particularly from patients with more severe asthma. IL-17-producing ILC3s can also be detected in human bronchoalveolar lavage fluid after in vitro stimulation with IL-2, IL-7, and IL-1β (23). This indicates that ILC3s are present in the lungs of patients with asthma and might play an important role in some forms of this disease.
Unlike the study of mice, the study of human has many limitations; however, a growing body of evidence indicates that ILCs are present in the human immune system. Therefore, more precise studies are needed, since ILCs seem to play an important role in the development of various immune diseases.
CONCLUSION
The discovery of innate lymphoid cells (ILCs) over the past several years has changed our understanding of the principles of immune regulation. ILCs have critical roles in the cytokine-mediated regulation of several diseases including autoimmune diseases, infectious diseases, and allergic diseases. Indeed, it is noteworthy that subsets of ILCs seem to be particularly prevalent at mucosal surfaces (e.g., respiratory tract, skin, and intestine), which are constantly exposed to infectious agents in the external environment. The key function of the epithelial barriers of the skin, respiratory tract, and intestine is to limit exposure to commensal and pathogenic microbes and maintain tissue homeostasis. However, in addition they are also involved in immune responses through the production of innate cytokines, which initiate the function of ILCs. ILCs might have evolved to play critical roles in host defense, regulation of inflammation, and tissue repair through the rapid production of large amounts of cytokines.
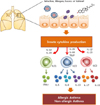
Studies of ILCs have opened new areas of investigation in allergic diseases and asthma. ILCs are thought to be the main producers of effector cytokines such as IL-5, IL-9, IL-13, IL-17, and IL-22, which are involved in the development of different forms of asthma (Fig. 1). ILC2s in particular play very important roles in the development of asthma by producing large amounts of type 2 cytokines (IL-5, IL-9, and IL-13) in response to IL-25, IL-33, and TSLP. In addition, ILC3s, at least under stress conditions (like obesity), seem to play a critical role in inducing asthma by secreting IL-17 in response to IL-1β. The roles of IL-22-producing ILC3s and of ILC1s in the development of asthma still need to be explored.
Although there have been many new findings regarding ILCs, many unanswered questions remain. However, it is certain that ILCs are important therapeutic targets in several respects. First, ILCs can regulate the adaptive immune response by early production of cytokines. Second, ILCs can interact with various cell types, such as natural killer T cells, mast cells, macrophages, eosinophils, as well as T cells, and regulate their function. Further investigation of this particular cell subset will lead to new ways of targeting many unresolved diseases.

 XML Download
XML Download