

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abbreviations
RHD
rel homology domain
TAD
transactivation domain
TCR
T cell receptor
PI3K
phosphoinositide 3-kiase
PDK1
phosphoinositide-dependent kinase 1
CBM
Carma1-Bcl10-Malt1
GLK
GCK-like kinase
Th
T helper
RA
rheumatoid arthritis
IBD
inflammatory bowel disease
SLE
systemic lupus erythematous
PGE2
prostaglandin E2
INTRODUCTION
NF-κB is activated during immune responses and is important for the expression of immune response related genes including cytokine, chemokine, and adhesion molecule genes (1-3). The NF-κB family is composed of RelA, RelB, c-Rel, p50 (NF-κB1), and p52 (NF-κB2) subunits. The NF-κB transcription factor binds to κB sites as dimers, either homodimers or heterodimers. The NF-κB protein contains N-terminal Rel homology domain (RHD), which makes contact with DNA and supports subunit dimerization. Of the NF-κB subunits, only RelA, RelB, and c-Rel have transactivation domain (TAD) at C-terminus and this TAD domain is important for initiation of target gene transcription (1-3). However, p50 and p52 lack TAD domain. Thus, p50 and p52 can positively regulate gene expression through heterodimerization with TAD containing NF-κB subunits or other regulators (Fig. 1).
NF-κB complexes are inactive in most cells, and these complexes are located in the cytoplasm in a complex with inhibitory IκB proteins (IκBα, IκBβ, IκBε, IκBζ p100, p105, Bcl3, and IκBns). NF-κB pathway activating signals including cytokine receptor signals and antigen receptor signals activate the IκB kinase (IKK) complex, which phosphorylates IκB. This phosphorylation induces IκB degradation, which leads to NF-κB complex translocation to the nucleus. Once in the nucleus, the NF-κB complex activates target gene transcription (1-3).
NF-κB PATHWAY IN T CELL ACTIVATION
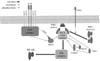
Antigen recognition by T cell receptor (TCR) induces activation of many transcription factors including NF-κB, NF-AT, and AP-1 (2,4), which are important for the induction of proliferation of activated T cells and their differentiation into Th1, Th2, Th17 and other Th cells (2,5). Ligation of CD28 co-receptor, along with the TCR, is essential for full activation of T cells (6,7). Especially, CD28 co-receptor ligation is required for efficient activation of NF-κB. It is well known that CD28 greatly enhances phosphoinositide 3-kinase (PI3K) activity and this activity is required for many cellular responses including cell survival, and cell proliferation (8). The CD28 cytoplasmic tail contains a PI3K binding motif such as YMNM motif (9). PI3K has been reported to bind to the YMNM phosphotyrosine, which leads to PI3K activation. CD28-mediated PI3K activation is involved in phosphoinositide-dependent kinase 1 (PDK1) and AKT recruitment into the immunological synapse (10-12), which activates PDK1 and AKT (13). These processes are important for PKCθ and Carma1-Bcl10-Malt1 (CBM) complex-mediated NF-κB activation during T cell activation (13-15). However, the concept of NF-κB activation by YNMN-mediated PI3K activation has been challenged because YNMN motif mutation had no significant effect on T cell proliferation and IL-2 production (16,17). The report suggested that another region of the CD28 cytoplasmic tail is responsible for PDK1 activation and subsequently this induces PKCθ-mediated NF-κB activation (17). A recent in vivo infection experiment suggested that another CD28 cytoplasmic tail region is responsible for NF-κB activation (10). Even though the exact CD28 cytoplasmic tail region for NF-κB activation is controversial, CD28-mediated PDK1 activation and subsequent PKCθ-mediated NF-κB activation pathway is well supported by previous studies (13,17,18).
During T cell activation, CD28 recruits PDK1 into the immunological synapse where the recruited PDK1 is converted into competent states for binding to down-stream molecules such as PKCθ and Carma1 (13,19). During this process, phosphorylation of threonine 513th on PDK1 is important for the conversion of PDK1 heterotypic dimer to homotypic dimer, which enables the formation of the PDK1-PKCθ-CBM complex. In addition, recently, GCK-like kinase (GLK) was suggested as the kinase for PKCθ phosphorylation at threonine 538th (20). Thus, it is possible that PDK1 works as a scaffold for the complex formation. Subsequently, IκB kinase (IKK) is activated by the signaling complex (2), and the IKK complex activates NF-κB during T cell activation (Fig. 2).
Th17 CELLS
CD4 T cells play an essential role in the adaptive immune response. During the adaptive immune response, activated CD4 T cells differentiate into T helper (Th) 1, Th2 or Th17 effector cells. Th1 cells are important in host defense against intracellular pathogens and Th2 cells are involved in allergic immune responses and defense against parasite infections. Th17 cells play an important role in host defense against extracellular pathogens and fungal infections (21,22). Furthermore, Th1 and Th17 cells are important in intestinal immune responses. During CD4 T cell differentiation, IL-6 and TGF-β are important for Th17 cell differentiation. IL-23 is also involved in Th17 cell differentiation (23). In addition to the direct effect of IL-23 on Th17 cell differentiation, IL-23 stimulates intestinal TCRγδ T cells, invariant natural killer T cells (iNKT), and intestinal innate-like T cells to secrete cytokines related to Th17 differentiation (24). In addition to TGF-β and IL-23, IL-6 is also important for Th17 differentiation. However, while TGF-β negatively regulates human Th17 cell differentiation, this cytokine is important for Th17 cell differentiation in murine Th17 differentiation (25-27). During Th17 cell differentiation, the transcription factors RORγt, RORγ, RORα, IRF4, and STAT3 are important for effector T cell differentiation. However, IFN-γ, IL-2, and IL-4 negatively regulate Th17 cell differentiation (28,29).
Th17 CELLS IN AUTOIMMUNE DISEASE
Differentiated Th17 cells produce proinflammatory cytokines such as IL-17A, IL-17F, IL-21, TNF, and GM-CSF (5,30,31). These cytokines are important in host defense against extracellular bacteria through acute immune responses (32). In addition, Th17 cells are involved in the development of autoimmune diseases including rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and multiple sclerosis (26,33).
It has been suggested that unbalanced immune responses can induce inflammatory diseases such as IBD. The detailed mechanism of IBD induction, including Crohn's disease and ulcerative colitis, has not been clarified (34-36); however, uncontrolled T cell activation and biased effector T cell (Th1, Th2, and Th17 cells) differentiation have been suggested as causative factors. Unbalanced production of Th17-related cytokines is also involved in the induction of IBD and other autoimmune disease. Th17 cells produce cytokines including IL-17A, IL-17F, IL-21, and IL-22 (37,38). IL-17 is the representative cytokine produced by Th17 cells and is involved in RA, asthma, and systemic lupus erythematous (SLE) development. A number of studies have investigated the role of IL-17A in intestinal inflammation, and showed that IL-17A is overproduced in patients with Crohn's disease and ulcerative colitis (39-42). In addition, IL-17 family cytokines are also increased in patients with autoinflammatory diseases including RA, asthma, and SLE (43-45). IL-17 and IL-23R genomic DNA sequence analysis found polymorphic regions related to IBD induction (46,47). In addition, IL-21 produced by Th17 cells was found to be involved in exacerbation of IBD (48,49). Furthermore, IL-21 gene deleted mice are resistant to Th1/Th17 cell-mediated colitis induction (50).
EXTRINSIC EFFECT OF NF-κB ACTIVATION ON Th17 CELL DIFFERENTIATION
The NF-κB pathway regulates antigen presenting cell functions and affects CD4 T cell differentiation into Th effector cells (51,52). Dendritic cells are the most important antigen presenting cells for Th cell differentiation. RelA deficiency reduced IL-1α, IL-1β, and IL-6 production from dendritic cells in response to LPS stimulation (53). In fact, these cytokines are involved in Th17 cell differentiation (25). In inflammatory responses, prostaglandin E2 (PGE2), an endogenous lipid mediator, enhances the production of IL-1β and TNF-α from bone marrow derived dendritic cells. In addition, PGE2 reduces the level of IL-12, but increases the level of IL-23 production. In addition to changes in cytokine production, PGE2 affects the expression of TLR-4, 2, and 9, IL-1R-associated kinase 1 (IRAK1), IKK, and p38 activator (MKK3), which are important components of the NF-κB activation pathway in dendritic cells. In addition, PGE2 reduces the level of IL-12, but increases the level of IL-23 production. In addition to changes in cytokine production, PGE2 affects the expression of TLR-4, 2, and 9, IL-1R-associated kinase 1 (IRAK1), IKK, and p38 activator (MKK3), which are important components of the NF-κB activation pathway in dendritic cells. Moreover, PGE2 treatment of bone marrow derived dendritic cells increases Th17 cell differentiation in vitro (54). Thus, it is suggested that NF-κB modulation in antigen presenting cells can also affect Th17 cell differentiation.
INTRINSIC EFFECT OF NF-κB ACTIVATION ON Th17 CELL DIFFERENTIATION
Binding of RORγt and RORγ to IL-17A and IL-17F promoters regulates their expression, which is important for Th17 differentiation. It has been shown that NF-κB subunits such as p65 and c-Rel are localized in the RORγt and RORγ promoter regions and affect RORγt and RORγ gene expression. Thus, it has been suggested that NF-κB activation is important for Th17 differentiation (55). Also, IKKβ can stimulate PKCθ-mediated STAT3 promoter activation. This promoter activation is essential for Th17 cells differentiation (56). During T cell activation, Malt1 is an important component of the NF-κB activation pathway through regulation of IKK activation. In in vitro conditions, naïve T cell differentiation into Th17 cells is decreased by Malt1 deficiency (57). In addition, Carma1, which is adaptor protein for TCR-mediated NF-κB activation, is needed for expressions of IL-17A, IL-17F, IL-21, IL-22, IL-23R, and CCR6. Carma1 deficiency also blocks Th17 cell development because chromatin loci of Th17 effector molecules cannot form open conformation, but transcription factors, which are needed for Th17 cells development, were normally expressed (58). In addition, overexpression of calpastatin minimal domain, which is a natural inhibitor of calpain, also decreased Th17 cell differentiation through stabilization of IκBα and subsequent inhibition of NF-κB activation (59). In addition, one recent report described an important role for IκBζ in Th17 differentiation, and showed that IκBζ acts with the nuclear orphan receptors RORγ and RORα to promote IL-17A gene expression (60). Thus, many data support the importance of NF-κB activation in Th17 cell differentiation.
However, recently, negative results were also reported. IL-2 is secreted from activated T cells, and acts as a negative regulator of Th17 cell differentiation (61). In the above report, c-Rel was suggested as a positive regulator of Th17 cell differentiation. c-Rel deficiency in T cells decreases IL-2 production from activated T cells. However, this condition did not affect Th17 cell differentiation. In the report, even though IL-2 was added in Th17 cell differentiation conditions, it did not affect Th17 cell differentiation. IRF-4 is a positive regulator of Th17 cell differentiation; however, it was not affected by c-Rel deficiency. Thus, the report argued that c-Rel is not involved in Th17 cell differentiation (62). It has been reported that USP18 regulates the TAK1-TAB1 complex, which is known as NF-κB pathway. USP18 deficient T cells showed NF-κB hyperactivation, and subsequently increased the level of IL-2 secretion. This dysregulation of NF-κB reduced Th17 cell differentiation (63).
CONCLUSION
NF-κB activation is important during T cell activation and for cytokine gene expression in antigen presenting cells. NF-κB activation-mediated Th17 cell cytokine gene expression is important for Th17 cell differentiation; however, different experimental systems showed different roles of antigen receptor-mediated NF-κB activation in Th17 cell differentiation (Fig. 3). Thus, deciphering the role of NF-κB in each of the Th17 cell differentiation conditions, such as different disease states, is an area of great interest.

 XML Download
XML Download