

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
MicroRNAs (miRNAs) are small non-coding RNAs of approximately 19 to 25 nucleotides that post-transcriptionally regulate gene expression. They bind to the 3'-untranslated region (UTR) of their target messenger RNAs (mRNAs) through complementary recognition, which then leads to mRNA degradation or repression of protein expression (1,2). To date, over 2000 mature miRNA products have been identified in the human genome and the number registered on the miRNA database is still growing. More important than the expanding numbers is the complex regulatory network mediated by miRNAs, which controls a spectrum of biological events ranging from cell differentiation, proliferation and homeostasis, cell-cell interactions to intracellular signalling responses (3,4,5,6). Furthermore, miRNAs play important roles in the development and function of innate and adaptive immune cells (7,8). Many immunoregulatory genes, including transcription factors, cofactors and chromatin modifiers, are miRNA targets and some even harbour binding sites for eight or more different miRNAs (9). Likewise, each miRNA could potentially recognise many, or even up to hundreds of, target genes (4). Therefore, dysfunction of miRNAs or dysregulation of their expression in the immune cells would lead to immunodeficiency or autoimmunity.
Systemic lupus erythematosus (SLE) is a potentially lethal chronic autoimmune disease, which affects multiple organ systems ranging from the skin, kidney, central nervous system, to the haematological, musculoskeletal, cardiovascular and the gastrointestinal systems (10,11,12). Overall, the array of clinical manifestations in SLE is intricate, suggesting that the pathogenesis of SLE may involve the interplay of multiple factors. Immunologically, SLE is the consequence of loss of self-tolerance and amplification of self-antigen-mediated hyperactivation of T and B lymphocytes. Since the discovery of miRNAs, a growing attention has been drawn to their involvement in autoimmunity (13,14,15). In this review, we will first briefly summarise the key features in miRNA biogenesis and how its dysregulation mediates autoimmunity. Recent researches on aberrant miRNAs expression in SLE and current insights on miRNA-mediated dysfunction of T and B lymphocytes and their contributions in the development of SLE will be discussed. Finally, we highlight the immunoregulatory role of miR-146a and its possible association with SLE pathogenesis.
MicroRNA BIOGENESIS AND AUTOIMMUNITY
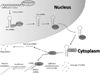
The biogenesis of miRNAs, as illustrated in Fig. 1, starts from the transcription of unique miRNA genes or intron regions of protein-coding genes by RNA polymerase II (2). The immediate transcription products are hairpin-structured primary miRNAs (pri-miRNAs) in various lengths. These pri-miRNAs are processed into precursor miRNAs (pre-miRNAs) that are generally 70 to 120 nucleotides long by the microprocessor complex containing the RNase III enzyme Drosha and the dsRNA binding domain protein DiGeorge Syndrome critical region 8 (DGCR8) (16). Subsequently, the pre-miRNAs are transported from the nucleus to the cytoplasm by exportin 5 (XPO5) (17). In the cytoplasm, the RNase Dicer excises the terminal loop of the miRNA so that it turns into an 18- to 24-nucleotide duplex. The duplex is then incorporated into the RNA interfering silencing complex (RISC), where it is unwound into single-stranded mature miRNA. The other strand, also called the star form of the miRNA, is usually degraded soon afterwards. The RISC is directed to the target mRNA via the complementary base-pairing between the miRNA and the mRNA. Often, the complementary base-pairing is not perfect and there are mismatches and bulges between the two strands. The fate of the target mRNA is believed to be dependent on the level of complementarity. When there is near perfect or sufficient complementarity, the target mRNA will be cleaved and degraded; while in cases of insufficient complementarity, the productive translation of the mRNA will be repressed instead (18).
The importance of miRNAs in the immune system and their association with autoimmune diseases have been demonstrated through in vitro and in vivo experiments by inactivating key enzymatic components in the miRNA biogenesis. By conditionally deleting Drosha or Dicer in total T cells or CD4+ T cells, it has been shown that miRNA biogenesis is indispensable for the function and homeostasis of mature T lymphocytes, particularly in the regulatory T (Treg) cell compartment (19,20,21). Despite having a reduced number in mature T cells, these mice spontaneously develop inflammatory diseases in conjunction with a compromised Treg suppressive activity, suggesting that these miRNA processing enzymes are critical for Treg function and hence self-tolerance maintenance (20). Indeed, mice with Dicer-deficiency specifically in Treg cells (22,23) are phenotypically similar to the Foxp3 knockout (KO) mice (24) and develop systemic autoimmune disease marked by lymphocytic and monocytic infiltrations in multiple organs, lymphadenopathy and splenomegaly. Similarly, spontaneous Dicer insufficiency in Treg cells has been observed in MRL/lpr lupus mice in association with a reduced suppressive activity (25), further indicating the critical involvement of miRNAs biogenesis in normal Treg function. In parallel, several studies have also reported the importance of miRNA biogenesis in B cell development and functions through conditional Dicer-KO in early B cell progenitors, CD19+ B cells or activated B cells (26,27,28,29). It is noteworthy that Dicer-deficiency in the CD19+ B cell compartment also promotes autoimmunity and the aged female mice exhibit a skewed antibody repertoire with significantly increased IgG titres against dsDNA, ssDNA and cardiolipin autoantigens, as well as augmented immune complex depositions in the kidneys (26).
In addition to being the cause of autoimmunity, dysregulated miRNA biogenesis may also be the consequence of autoimmune conditions. Intensified oxidative stress, elevated levels of proinflammatory cytokines and autoantibodies could affect the expression of miRNAs by interfering key components of the miRNA biogenesis machinery (30,31,32). For instance, treatment of H2O2 and type I interferon (IFN) post-transcriptionally represses protein level of Dicer and leads to miRNAs differential expression in three different human cell lines (30). In addition, the autoreactive anti-Su antibodies in sera of patients with systemic rheumatic diseases has been found to cross-react with argonaute 2 (Ago2), the catalytic core enzyme in the RISC complex, and potentially affecting the miRNA synthesis in autoimmune conditions (31,32). Taken together, interrupted miRNA biogenesis strongly associates with autoimmunity and possibly plays a role in promoting disease progression through reciprocal interactions between the major processors of the miRNA pathway and their targets.
DYSREGULATED microRNA EXPRESSION PROFILES IN SLE
Comprehensive analysis on the expression profiles of miRNAs have revealed intriguing patterns and shown to be beneficial in human cancer research (33). Similarly, studies on immune cell-derived or circulating miRNAs expression profiling in patients with SLE is likely to provide useful information for understanding SLE pathogenesis or for developing prognostic biomarkers and novel therapeutics. Table I summarises several reports from 2007 to 2013 on systematic analysis of miRNA profiling in SLE patients in comparison with healthy individuals. Collectively, there is a lack of distinct pattern in specific dysregulated miRNA expression in SLE among the reviewed studies. Variations in the ethnicity of research subjects, the sizes of screened population and the types of biological sample tested as well as the detection methods could contribute to the discrepancies in the miRNAs identified in different studies. For instance, SLE serum miR-223 level was found to be increased in the study by Wang and colleagues (34), but significantly decreased in patients with active lupus nephritis in the study by Carlsen et al. (35). Notably, the latter study collected patient samples mostly from the European ancestry while the former one from the Chinese population. Potentially, biological samples collected from different ethnic groups, patients recruited at different disease stages or receiving different treatments could yield alternative expression profiles of miRNAs.
Despite the lack of consistency in specific dysregulated miRNAs among the profiling studies, there are commonalities in the effector or functional outcomes resulting from the altered miRNA expression. Firstly, miRNAs with the greatest change of expression in peripheral blood mononuclear cells (PBMCs) from lupus patients may target the key components of a common pathway, the type I IFN signalling cascade (36,37). The under-expression of miR-146a has been found negatively correlated with SLE disease activity and IFN scores (36). At the molecular level, miR-146a targets IRF5 and STAT1 (36) which are important transcription factors related to IFN signalling. In a separate study, bioinformatics analyses have revealed several IFN signalling mediators and IFN-inducible genes as the potential targets of five miRNAs with major changes in SLE PBMCs and lupus patients-derived cell lines (37). Type I IFN is recognised as the central player in SLE pathogenesis from the striking correlation between the dysregulated type I IFN-inducible gene expression pattern and SLE disease activity, also known as the "IFN signature" of SLE (38,39,40). Therefore, the commonly affected IFN-related pathway indicates the essential roles of distinct groups of miRNAs in SLE pathogenesis. Secondly, profiling studies on PBMCs as well as purified T cells have also highlighted the significance of miRNA dysregulation leading to T cell hyperactivity in SLE (41,42,43,44). Functional studies on lupus T cells further reveal common regulatory patterns by various miRNAs (Table I) and this will be discussed in the later section.
Overall, although it is difficult to identify a stable, specific and sensitive "miRNA signature" for SLE at the current stage, it may be possible to pinpoint distinct groups of dysregulated miRNAs according to their shared functional consequences in SLE. Together with advance technologies, systematic and stringent sample selection, followed by independent validation of individual miRNAs, a more representative disease-related miRNA profile should promote a better diagnostic and/or therapeutic use in SLE.
MicroRNA REGULATES T CELL FUNCTIONS IN SLE
In SLE, miRNA-mediated dysregulation has been studied mostly in T cells, one of the major cellular effectors of the disease. Many of these studies focus on the mechanism(s) mediated by individual miRNA which effects in T cell hyperactivity or abnormality. In general, there are several ways that various miRNAs bring about the regulatory effect co-operatively. Notably, different dysregulated miRNAs can target the same molecules to maximize inhibition efficiency. For instance, miR-126 and miR-148a have been found up-regulated in SLE T cells, and albeit binding to different regions, they both directly target DNA methyltransferase 1 (DNMT1) (43,45), the key methyltransferase for maintaining DNA methylation during replication. Indeed, lupus patients could have a reduced DNMT1 expression (46) and an abnormal global DNA hypo-methylation in T cells, which is a major epigenetic trait in SLE (46,47,48). Remarkably, DNMT1 also serves as an excellent example to illustrate how various miRNAs act in concert on different molecular targets to achieve the same effector outcome(s). Overexpression of miR-21, miR-148a (45), miR-126 (43) and miR-29b (49) have been independently reported to positively correlate with DNA hypomethylation in lupus CD4+ T cells through the inhibition of DNMT1. The suppression is mediated either by directly targeting DNMT1 mRNA protein coding region or 3'-UTR (43,45), indirectly through the suppression of its transactivator Sp1 (49), or even further upstream of DNMT1 through regulation of RASGPR1 via the Ras-MAPK pathway (45). In these studies, the effector outcomes were the increased expression of CD11a and CD70 (43,45,49), both of which are methylation sensitive genes associated with SLE pathogenesis (48), and contributed to the exaggerated T cell responses such as T-cell assisted IgG production by plasma cells (43). Thus, these miRNAs may regulate other lupus-associated and methylation-sensitive components which are yet to be identified. Significantly, suppression of miR-21, miR-148a and miR-29b in lupus T cells could restore the normal phenotypes to a certain extent, further indicating their pathogenic involvement in SLE (45,49). Among the up-regulated miRNAs associated with SLE, miR-21 has been identified in several studies (44,45,50) and a strong correlation with SLEDAI score has been reported (44). In T cells, miR-21 regulates multiple pathways that lead to overall T and B cell hyperactivity. Apart from DNMT1 as described above, miR-21 also directly suppresses the selective protein translation inhibitor PDCD4 expression, leading to an enhanced proliferation, increased IL-10 and CD40L expression in lupus CD4+ T cells, and in turn promoting the differentiation of plasma cells and IgG production (44). Similar regulatory pathway has been observed in macrophages where miR-21-mediated suppression of PDCD4 favors the production of IL-10 upon TLR-4 stimulation via NF-κB pathway (51), supporting the idea that miR-21 may promote inflammation through suppression of negative regulatory mediators.
The perturbed lupus T cell functions are regulated by the synchronized down-regulation of several specific miRNAs too. It is interesting to note that the reduced expression of miR-142 in lupus CD4+ T cells mediates similar outcomes as in the case of miR-21 overexpression. Reduced miR-142-3p/5p expression in SLE CD4+ T cells has been found inversely correlated with the expression of their putative targets including SLAM-associated protein (SAP), CD84 and IL-10 (52). Functionally, inhibition of miR-142-3p/5p in normal CD4+ T cells leads to over-activation as marked by elevated levels of IL-4, IL-10, as well as CD40L and ICOS protein expression. These miR-142-3p/5p-suppressed T cells could further promote B cell hyperactivity resulting in a higher IgG production while the miR-142-3p/5p-transfected lupus CD4+ T cells exhibit the reverse phenotypes (52). Although it is not clear whether SAP and CD84 are the direct molecular targets of miR-142, they are important components for optimal T- and B-cell interaction and are critical for productive IgG response (53). The essential role of SAP in lupus has been demonstrated in SAP-mutated MRL/lpr mice which display a repressed development of autoantibodies, splenomegaly and lymphadenopathy (54).
The aberrant production of cytokines and chemokines in SLE T cells are also mediated by the abnormal down-regulation of miRNAs. T cells from SLE patients produce remarkably low amounts of IL-2 upon activation (55), which is crucial for Treg cells maintenance (56). Many regulatory factors, including miRNAs, contribute to the reduced IL-2 production in lupus patients. A recent report has demonstrated that the significant reduction in miR-31 expression in SLE T cells is positively correlated with the lowered IL-2 production (57). While transfection of miR-31 mimics increases the production of IL-2, the knockdown of endogenous miR-31 reduces IL-2 expression in primary T cells. Further bioinformatics analyses and luciferase reporter assays have revealed that miR-31 could enhance IL-2 promoter activity through suppressing the expression of RhoA (57) and the kinase suppressor of ras 2 (KSR2) (58). The former is a small GTPase that negatively regulates nuclear factor of activated T cells (NFAT) for IL-2 transcription (59), and KSR2 is a repressor factor of Ras2 kinase which is an upstream component in T cell activation (58). Interestingly, miR-31 (and also miR-125a) has been identified as a signature miRNA for human natural (n)Treg cells due to its specific down-regulated expression (60). In vitro studies have shown that miR-31 negatively regulates FOXP3 expression by binding directly to its potential target site in the 3'-UTR, and that the overexpression of miR-31 in nTreg cells can lead to suppression in FOXP3 expression. Whether through down-regulation of IL-2 production or FOXP3 suppression, the physiological role of miR-31 in Treg maintenance in SLE, however, requires further investigation. Likewise, the role of miR-125a down-expression in nTreg cells is not clear. Yet, its under-expression in SLE CD4+ T cells is apparently affecting CC chemokine ligand 5 (CCL5, also known as RANTES), an inflammatory chemokine whose level was found elevated in sera of SLE patients (61,62). Zhao et al. showed that the reduced miR-125a in lupus PBMCs was mainly contributed by CD4+ T cells and was associated with an increased in Kruppel-like factor 13 (KLF13) (61), a key component of a regulatory complex controlling the expression of RANTES in T cells (63). Significantly, a rescued expression of miR-125a in lupus CD4+ T cells by transfection could alleviate the elevated RANTES expression, which likely resulted from the direct targeting of the 3'-UTR of KLF13 (61).
MicroRNA REGULATES B CELL FUNCTIONS IN SLE
Direct damages caused by autoantibodies and immune complexes in the inflamed tissues are the characteristic features in SLE. Long-lived and hyperactive B cells are the major sources for autoantibodies. Unlike T cells, there are fewer studies that systematically report miRNA dysregulation in lupus B cells. Liu and colleagues have reported a direct interaction between miR-30a and the 3'-UTR of Lyn mRNA in association with dysregulated B cell functions in SLE patients (64). Lyn, a key negative regulator of B cell activation, is significantly down-regulated in lupus B cells (65) and the defective Lyn expression is associated with B cell spontaneous proliferation and anti-dsDNA autoantibody production (66). Indeed, the dramatic increase of miR-30a expression in CD19+ B cells from SLE patients negatively correlates with the expression of Lyn, and the miR-30a overexpression increases B cell proliferation and IgG production through the inhibition of Lyn (64). Similarly, miRNAs may also affect functions of activated B lymphocytes by interacting with another critical enzyme, the activation-induced cytidine deaminase (AID) which initiates and regulates B cell secondary antibody diversification through somatic hypermutation and class switch recombination (67,68). AID-deficiency in the lupus-prone MRL/lpr mice has led to a complete reduction of anti-dsDNA IgG and a significant increase in animal survival with delayed nephritis development (69), indicating the pathological roles of AID in SLE. Subsequent studies have revealed that miR-155 and miR-181b are the negative regulators of AID (70,71,72), both of which have conserved binding sites in the 3'-UTR of AID mRNA. Overexpression of miR-181b in activated B cells leads to down-regulated mRNA and protein levels of AID (72). Splenic B cells from mice with disrupted miR-155-binding site in AID mRNA have a faster and higher AID expression in response to LPS and IL-4 stimulation (71). In addition, the induction of IgG3, IgG1 and IgG2a upon LPS, LPS with IL-4 or LPS with IFNγ respectively indicated an increased class switching in B cells from these mice in vitro. Interestingly, despite its negative regulatory role on AID, miR-155 has been found upregulated in splenic B cells from MRL/lpr lupus mice (73) and deletion of miR-155 in the B6-Faslpr mice results in a reduction of class switched IgG antibodies (74). Since there are fewer studies on the expression or function of miR-181b in lupus B cells, it remains unclear how miR-155 and miR-181b, and possibly other miRNAs, synergistically regulate the function of AID, leading to the development of SLE. Recently, a study using IFN-accelerated lupus mouse model has revealed the specific effect of miR-15a in regulatory B cell subsets and autoantibody production (75). In this study, IFNα administration to the NZB/W F1 lupus mice could promote proteinuria development in association with an increase in splenic and plasma miR-15a expression, which in turn correlated positively with anti-dsDNA antibody level. Intriguingly, there was a concomitant decrease in immunosuppressive B-10 cells which expressed high level of miR-15a. In addition, miR-15a expression in conventional B-2 cells increased and superseded the level in regulatory B-1 cells as the disease progressed, suggesting that miR-15a may have a regulatory role in balancing different B cell subsets. However, the mechanism involved is yet to be identified.
MiR-146a IN SLE
MiR-146a is a well-recognized negative regulator in autoimmunity. Mice deficient in miR-146a manifest severe auto immune phenotypes with elevation of autoantibodies, splenomegaly, lymphadenopathy as well as exaggerated immune responses towards LPS challenge (76). In SLE, miR-146a dysregulation is associated with perturbed IFN responses. In patients, miR-146a down-regulation in PBMCs negatively correlates with disease activity and IFN scores as marked by the elevated expression of signature genes such as Ly6E, OAS1 and MX1 (36). IFN regulation by miR-146a in PBMCs is likely mediated by direct interference with two IFN-related transcription factors IRF5 and STAT1. On the other hand, miR-146a has also been shown to negatively modulate type-I IFN production in macrophages through targeting TLR signaling molecules TRAF6, IRAK1 and IRAK2 (77). Interestingly, TLR7/9 stimulation in normal PBMCs induces miR-146a expression (36), which likely serves as a negative feedback loop for normal IFN response. It is possible that such negative feedback mechanism is impeded in SLE. A recent genetic analysis has identified a novel genetic variant (rs57095329) in the promoter region of miR-146a which confers association with its expression level (78). The SLE risk-associated G allele of the variant is linked to a reduced miR-146a expression in PBMCs, possibly by lowering the protein binding affinity and activity of the promoter. Further in vitro analyses have revealed a reduced binding of ETS-1, another SLE-susceptibility associated transcription factor, to the risk variant, suggesting that the combined effect of at least two risk factors may contribute to the reduced miR-146a expression in SLE (78). MiR-146a is often bundled with TLR7/9 stimulation, which plays a central role in SLE pathogenesis. Apart from PBMCs, TLR7/9 stimulation in plasmacytoid dendritic cells (pDCs) also induces miR-146a expression (79). Of relevance, despite its scarcity in peripheral blood, pDCs have been implicated for an essential role in SLE disease amplification by its unique ability to produce large amount of type-I IFN upon TLR7/9 stimulation (80). Two recent studies have indeed suggested miR-146a as a key regulator in pDC survival and functions. First, lentivirus-transduced overexpression of miR-146a in the CAL-1 pDC cell line has resulted in increased cell apoptosis, impaired TLR7-induced co-stimulatory molecules, IFN-β and IL-6 expression, as well as decreased capability of pDCs to drive CD4+ T cells proliferation (79). In another study, Charrier et al. have observed a much higher expression of miR-146a (and miR-155) in human umbilical cord blood (UCB) pDCs when compared to adult blood pDCs (81). This higher miR-146a expression in UCB pDCs was associated with a reduced expression of TLR9/IRF7 pathway proteins, namely TLR9, MyD88, IRAK1 and IRF7, and in turn resulting in a lower production of IFN-α upon TLR-9 activation. Taken together, miR-146a likely plays a negative modulatory role in pDC survival and functions. Interestingly, we have previously reported a significant increase in frequency of pDCs with enhanced T cell stimulatory activity in SLE patients (82,83). Whether miR-146a down-regulation is involved in the pathogenic development of pDC abnormalities in SLE is yet to be verified.
CONCLUSION
Since the discovery of miRNAs, they have caught immediate attention for their fundamental role in fine-tuning immune cell development as well as adaptive and innate immune responses. To date, majority of the studies have highlighted the regulatory mechanism(s) of specific miRNAs in contributing to cellular dysfunction in SLE. The therapeutic potential of miRNA modulation has also been tested in some animal models of lupus. In BXSB lupus-prone mice, miR-146a restoration by a novel recombinant virus-like-particles (VLPs) approach has been shown to ameliorate SLE progression (84). MS2-miR-146a VLPs therapy has led to a drastic reduction in autoantibodies, total IgG and proinflammatory cytokines levels in these mice. Conversely, in vivo silencing of miR-21 by a tiny-targeting locked nucleic acid (LNA) nucleotides approach has also yielded favourable outcomes in the B6.Sle123 lupus mice. For instance, the splenomegaly can be reversed and the expression of the miR-21 target, PDCD4, as well as the frequency of Fas-expressing lymphocytes in these lupus mice were also decreased (85). Conceptually, lupus disease modulation by miRNA manipulation is feasible but the potential application in human awaits more evidence in the clinical settings. As the Sanger miRBase sequence database expands rapidly, comprehensive bioinformatics tools for miRNAs is needed to facilitate the analysis of the highly intricate regulatory networks. "MiRNomics" have already been applied in advanced investigation on cancer research as well as autoimmune diseases like multiple sclerosis (86). While characterisation of miRNA expression patterns in SLE patients can be of potential diagnostic use, discoveries in cell type-specific miRNA expression profile during disease progression may provide a further understanding for SLE immunopathogenesis. Indeed, a recent study has examined the miRNomes of mouse bone marrow-derived DCs at different stages of development, maturation and differentiation, and eventually pinpointing miR-30b as a negative regulator in immune responses (87). Although there is not a miRNomic study specifically on SLE pathogenesis yet, tracking the spatial and temporal regulation of miRNA expression along with lupus development seems to be an attractive approach to facilitate further understanding of such a complicated disease. Hopefully, notions gathered in this review could provide clues for future investigations on SLE aetiopathology as well as novel miRNA-based therapies.

 XML Download
XML Download