

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Abbreviations
IL
interleukin
TNFα
tumor necrosis factor-α
IBD
inflammatory bowel disease
NOD
nucleotide oligomerization domain
PBMCs
peripheral blood mononuclear cells
MDP
muramyl dipeptide
CD
Crohn's disease
SEMFs
subepithelial myofibroblasts
IFNγ
interferon-γ
IL-32γ-TG
IL-32γ transgenic mouse
WT
wild type
DSS
dextran sodium sulfate
DCs
dendritic cells
RA
rheumatoid arthritis
sRANKL
soluble receptor activator of nuclear factor kappa-B ligand
OA
osteoarthritis
Syk
spleen tyrosine kinase
JNK
C-Jun N-terminal kinase
FLS
fibroblast-like synoviocytes
siRNA
small interfering RNA
TSLP
thymic stromal lymphopoietin
TLR
toll-like receptor
BLP
bacterial lipoprotein
LPS
lipopolysaccharide
poly I:C
polyriboinosinic polyribocytidylic acid
dsRNA
double-stranded RNA
DAMPs
damage-associated molecular patterns
PR3
proteinase 3
INFLAMMATORY BOWEL DISEASE (IBD)
IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 for inflammatory cytokine production in peripheral blood mononuclear cells (PBMCs) (1). The activation of mucosal immunity requires nonspecific innate signals by various bacterial products via pattern-recognition receptors. IL-32 activity is enhanced by the intracellular NODs. The synergistic effect of IL-32 and NOD2 ligand synthetic muramyl dipeptide (MDP) on inflammatory cytokine productions is abolished in PBMCs of Crohn's disease (CD) possessing 3020insC mutation (1). This in vitro synergism between IL-32 and NOD2 ligand MDP is associated with high expression of IL-32 in human colon epithelial tissues. In addition, IL-32 synergizes with synthetic ligand of NOD1 FK-156 on cytokine productions but the effect is absent in NOD1-deficient macrophages (1). These results suggest that IL-32 and NODs pathway has important role in mucosal immunity.
Imaeda et al. has identified a new IL-32 isoform from human colonic subepithelial myofibroblasts (SEMFs). The new IL-32 isoform is named IL-32ε and lacks exon 3 and 4 of the longest IL-32γ isoform. The transcript of IL-32ε is significantly elevated in the inflamed mucosa of IBD patients. TNFα induces transcript of new IL-32ε in a dose and time dependent manner (2). Interestingly, stable transfection of IL-32ε significantly decreased TNFα-mediated IL-8 transcript in HT-29 cells, but the expression of IL-32α, shortest isoform lacking exon 3 and 7, has no effect on TNFα-mediated IL-8 transcript. Whereas, other study has shown that the level of IL-32α protein and mRNA transcript are evaluated in inflamed epithelial mucosa of IBD patients compared to colonic epithelial cells of normal individuals (3). With intestinal epithelial cell lines, the expression of IL-32α transcript and protein is increased by IL-1β, interferon-γ (IFNγ) and TNFα. TNFα plus IFNγ exert synergistic effect on IL-32α expression and also IL-32α is highly expressed particularly in epithelial cells of IBD and CD patients. In the ileal tissues of patients with AS and intestinal chronic inflammation, significant up-regulation of IL-32 levels was found as compared with non-inflamed AS patients and controls (4). Further studies suggested that the biological activity of IL-32 plays important roles through interaction with other inflammatory cytokines such as TNFα, IL-1β, and IFNγ in the pathophysiology of IBD and CD (5,6,7).
The function of IL-32 in intestinal inflammation is investigated in vivo experiment by using IL-32γ transgenic mouse (IL-32γ-TG) expressing human IL-32γ in mouse. Although IL-32γ-TG mice are healthy, constitutive serum and colonic tissue levels of TNFα are increased. Compared with wild type (WT) mice, IL-32γ-TG exhibited a modestly enhanced acute inflammation early following the initiation of dextran sodium sulfate (DSS)-induced colitis (8). However, after day 6, there is less colonic inflammation and improved survival rate compared with WT mice. Associated with attenuated tissue damage, the colonic level of inflammatory cytokine is significantly reduced in IL-32γ-TG-treated with DSS and also constitutive level of IL-32γ itself in colonic tissue is decreased (8). These results suggest that IL-32γ emerges as an example of how innate inflammation worsens as well as protects intestinal integrity.
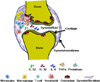
Fig. 1 illustrates induction of IL-32 from mucosal epithelial cells after infection of pathogens. IL-32 stimulates monocytes for inflammatory cytokines as well as differentiates monocytes into macrophage or dendritic cell (DC) like (9). Also IL-32 directly stimulates neutrophils to produces IL-6 and IL-8 (8,10,11). The differentiated macrophages and DCs are potent producers of key inflammatory cytokines in IBD and CD such as TNFα, IL-1β, and IL-6. These inflammatory cytokines in the inflamed area recruit T-cells, which are proliferated by the differentiated DCs to protect a host against the pathogens. On the other hand, increased numbers of various immune cells in the absence of proper immune suppressor molecules induces infiltration of neutrophil population in the inflamed area resulted in releasing a large amount of neutrophil proteinase such as elastase, proteinase 3 (PR3), and cathepsin G. These serine proteinase family enzymes are strong mediators of mucosal tissue damage exacerbating inflammation in IBD and CD. Although IL-32 expressions are elevated in inflamed mucosa epithelial cells of IBD and CD patients the biological activity of IL-32 in vitro and in vivo is inconsistent. Eight IL-32 mRNA transcripts generate five IL-32 isoform proteins (unpublished data). The discrepancy of in vitro and in vivo data could be because each investigator has studied a distinct IL-32 isoform or the regulation and function of IL-32 is complexity. Further studies are necessary to evaluate the precise function of IL-32 in IBD and CD.
RHEUMATOID ARTHRITIS (RA)
The effects of the most biologically active IL-32γ isoform on the differentiation of osteoclasts and IL-32 expression in rheumatoid arthritis (RA) have been investigated. Monocytes CD14+ from healthy volunteers or RA patients as well as synovial tissue of RA have been used to investigate the role of IL-32 in RA. The levels of IL-32γ are elevated in RA patients and IL-32 exacerbates mice models of experimental inflammatory arthritis (11,12,13). The osteoclastogenic effect and resorbed area are enhanced in the presence of soluble receptor activator of nuclear factor κ-B ligand (sRANKL) and the effect is more significant in the IL-32γ-treated cultures than that of IL-17 (11). The data suggested that IL-32γ is a potent mediator of active osteoclast generation in the presence of sRANKL. IL-32 is highly expressed in RA synovial tissue biopsies, whereas IL-32 was not observed in synovial tissues from patients with osteoarthritis (OA) by immune staining (14). The level of IL-32 expression is correlated with erythrocyte sedimentation rate, a marker of systemic inflammation. IL-32 is a potent inducer of prostaglandin E2 release in mouse macrophages and human blood monocytes. In TNFα-deficient mice, IL-32-driven joint swelling is absent and cell influx is markedly reduced suggesting that IL-32 activity is TNFα-dependent in RA (14).
Moon et al., has investigated extensively the signal pathway of TNFα-mediated IL-32 induction and they have characterized that TNFα-induced IL-32 is regulated through the spleen tyrosine kinase (Syk)/protein kinase Cδ (PKCδ)/c-Jun N-terminal kinase (JNK) pathways in RA synovial fibroblasts (12). IL-32 is elevated in fibroblast-like synoviocytes (FLS) from RA, whereas not in OA. TNFα-mediated IL-32 expression is specifically suppressed by inhibitors of Syk, PKCδ, and JNK as well as by small interfering RNA (siRNA) of these kinases (12). The levels of IL-32 and TNFα in the active RA groups are higher than those in the stable RA and control groups and also IL-32 level is positively correlated with other inflammatory markers in RA (15). IL-32 increases thymic stromal lymphopoietin (TSLP) production in human monocyte THP-1 cell line and PBMCs. IL-32 induces the differentiation of monocytes via TSLP since the blockade of TSLP prevents the monocytes differentiation into macrophage-like cells (16). Gene expression in cultured FLS from RA (RA-FLS) has been compared with gene expression in cultured FLS from OA (OA-FLS) using microarray analysis and IL-32 is the most prominently differentially expressed gene with higher expression in RA-FLS than in OA-FLS (17).
IL-17 induces IL-32 expression in the FLSs from RA patients and conversely IL-32 in the FLSs from RA patients stimulates IL-17 production from CD4+ T cells. Unlike the previous report (11), IL-32 and IL-17 synergistically induces the differentiation of osteoclasts. IL-32 and IL-17 also could induce resorption by osteoclasts in a RANKL-dependent manner. Both IL-32 and IL-17 can reciprocally influence each other's production and amplify the function of osteoclastogenesis in the in RA synovium. IL-32 and IL-17 separately stimulated osteoclastogenesis without RANKL and IL-32 synergistically amplified the differentiation of osteoclasts in the presence of IL-17, which is independent of RANKL stimulation. These data are similar to the result of IL-32 on osteoclastogenesis, but the co-stimulatory effect of RANKL different from previous report (11).
The serum level of IL-32 was assessed by using a clinical study with anti-TNFα therapy. At 24 weeks of treatment, serum samples of etanercept (also known as Enbrel, TNF binding protein) plus methotrexate responders had decreased IL-6 whereas increased IL-32 and IL-21. However, there were no differences in cytokine levels in non-responders (18). Pro-inflammatory cytokines contribute to persistent in chronic inflammation of RA and Etanercept therapy regulates level of serum cytokines. Interestingly, the serum level of IL-32 and IL-21 is specifically increased in etanercept responders. In contrary, treatment of RA patients with anti-TNFα significantly decreases IL-32 in synovial tissue (19). TNFα potently induces IL-32γ expression in FLS and the elevated TNFα, IL-1β, IL-6 and CXCL8 (also known as IL-8) productions are detected after IL-32γ overexpression in the presence of LPS in THP-1 cells. TNFα stimulation of FLS after IL-32γ/siRNA decreases IL-6 and CXCL8 production, whereas IL-32γ overexpression enhances IL-6 and CXCL8 (19). Additional studies are necessary to resolve the inconsistency of IL-32 expression in RA patients.
The overexpression of splice-resistant IL-32γ mutant in THP1 cells or RA synovial fibroblasts increases an important pro-inflammatory cytokine IL-1β compared with IL-32β (20). The result suggests that splicing to one less active IL-32β appears to be a salvage mechanism to reduce inflammation. Also the overexpression of primarily IL-32β in RA synovial fibroblasts decreases IL-32β secretion resulting in less inflammatory cytokine production. IL-32β lacks exon 3 possessing 46 amino acids, which contains a weak signal peptide of IL-32γ isoform whereas the overexpression of splice-resistant IL-32γ mutant in RA synovial fibroblasts enhances IL-32γ secretion. In addition, the level of TNFα and IL-6 production is associated with IL-32γ level in RA patients. These data reveal that naturally occurring IL-32γ, the longest isoform with the greatest activity among five IL-32 isoforms (10), can be spliced into IL-32β, which is a less active proinflammatory mediator.
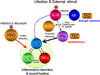
Toll-like receptor (TLR)-2, -3, and -4 ligands as well as IFNγ and TNFα induces IL-32β, γ and δ mRNA expression by RA FLSs (21). Mature IL-32 is expressed intracellularly and released by cells stimulated with the various activators. The IL-32α isoform was expressed intracellularly in response to TNFα and polyriboinosinic polyribocytidylic acid (poly I:C) and not released in culture supernatants. Stimulation of FLS with TNFα, bacterial lipoprotein (BLP), lipopolysaccharide (LPS), or poly I:C concomitant with IFNγ increases IL-32 expression compared with stimulation with IFNγ alone. IL-32 synthesis by FLSs is tightly regulated by innate immunity in RA. Therefore, TNFα, IFNγ, double-stranded RNA (dsRNA), hyaluronic acid, or other damage-associated molecular patterns (DAMPs) secretion in synovial tissues of RA patients may trigger IL-32 expression in RA patients. In inflamed synovial spaces, various infiltrated immune cells producing inflammatory cytokines such as TNFα, IL-1β, and IL-6 stimulates FLS to induce IL-32 and also DAMPs from death cells synergies with IL-32 further enhancement of inflammatory cytokine productions (Fig. 2).
CONCLUSION
The regulation of IL-32 is described in Fig. 3. Initial discovery of cytokine IL-32 was identified with in vitro experiment of microarray by using A549/IL-18Rβ stable cells that is indicated by blue arrow in Fig. 3 (22). However, the major route of IL-32 induction in vivo is probably downstream of IFNγ. Helminth antigen drives Th2 immune response via IL-18 alone whereas virus, M. Tuberculosis, and M. Leprae infection derive Th1 immune response via IL-12 plus IL-18 pathway. Activated T-cells and natural killer cells produce a large amount of IFNγ. Single and double stranded viral RNA in the presence of IFNγ are strong inducers of IL-32 in vivo and in vitro. In the other hand, infection directly activates neutrophils producing PR3, which is a mast regulator of IL-32, TNFα, and IL-1β. The innate and acquired immunity derived chronic local inflammation may contribute to IL-32-associated inflammatory disorders or wound healing process (Fig. 3). IL-32 is involved in both tissue damage and wound healing in the diseases, but further studies are necessary to resolve specific mechanisms of the reciprocal processes.

 XML Download
XML Download