

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Rheumatoid arthritis (RA) is a chronic inflammatory disease primarily that affects synovial tissues and results in the destruction of the affected joints (1). A variety of activated inflammatory cells, including innate and adaptive immune cells, appear to be involved in the destruction of the cartilage (1,2). Although the pathogenesis of this disease is multifactorial, RA susceptibility and severity have been associated with the presence of certain HLA class II alleles (3,4), which identifies T cells as an important mediator in the pathological processes underlying RA (5). T-helper cell phenotypes of RA typically exhibit a Th1-mediated response, and these T cells contribute, either directly or indirectly, to the proinflammatory cascade, in which cytokines play an essential role (3,6). Among these cytokines, tumor necrosis factor-α (TNF-α) has been implicated as the principal mediator of synovitis, and the neutralization of TNF-α activity results in an amelioration of RA-associated inflammation (7-9). In addition to TNF-α, inflammatory cytokines, such as IL-1, IL-6, IL-15, and IL-18, have also been reported to exacerbate the disease process, whereas IL-10 has been shown to suppress arthritis in several animal models (9-11). Recently defined Th17 cells and IL-17 they produce have been highlighted in the pathogenesis of RA (12-15). IL-17 induces production of inflammatory cytokines and mediators, such as IL-6, IL-1β, TNF-α, and PGE2, from synovial fibroblasts and macrophages and promotes expression of adhesion molecules (16). In addition, IL-17 induces expression of a TNF superfamily member, receptor activator of nuclear factor-κB ligand, which promotes osteoclast differentiation and leads to bone destruction (14).
CP-690550 is a newly developed inhibitor of JAK that has been shown to have the highest specificity and efficacy for JAK3 kinase inhibition in vitro relative to other known JAK3 inhibitors (17). Furthermore, CP-690550 prolongs allograft survival in murine and primate models with single mode therapy without any need for combined treatment with other immunosuppressants (17-19). Prolongation of allograft survival by CP-690550 operates through reduction in the numbers of T and NK cells and inhibition of effector cytokine production (20). In our previous report, CP-690550 treatment during the initial period of disease induction showed long-term disease protection in an allogeneic murine acute graft-versus-host disease (GVHD) model through preferential inhibition of IFN-γ-producing Th1 differentiation (21). A recent report also suggested that CP-690550 modulates both innate and adaptive immune responses affecting production of inflammatory mediators and T-helper cell differentiation (22). In addition to transplantation and associated GVHD, treatment window of CP-690550 has been extended to autoimmune inflammatory diseases, such as RA and psoriasis (23-26). Thus, it is meaningful to evaluate the long-term effect of CP-690550, in a murine model of arthritis more relevant to human RA as shown in a GVHD model.
In this study, we investigated the long-term therapeutic effect of CP-690550 on the pathogenesis of SKG arthritis, a animal disease model showing similar immunological, histopathological, and clinical characteristics with human RA (27). CP-690550 treatment remarkably suppresses disease progression in an SKG arthritis model when administered after the disease progresses to moderate-to-severe joint inflammation (score 3.5~4.0), and suppression of disease continues after cessation of CP-690550 treatment. Amelioration of disease severity correlates with reduced levels of pro-inflammatory cytokines, including IL-6, IL-17, and IFN-γ, and increased levels of IL-10 in joint tissue compared to a vehicle-treated control. Inflammatory cell infiltration in joint tissue and cavity is also decreased in CP-690550-treated mice compared to a vehicle-treated control. Thus, CP-690550 treatment has therapeutic effects on established SKG arthritis, and these effects continue after cessation of the treatment.
MATERIALS AND METHODS
Mice
SKG mice in a BALB/c genetic background were kindly provided by Dr. Shimon Sakaguchi (Department of Experimental Pathology, Institute for Frontier Medical Sciences, Kyoto University, Kyoto, Japan). Female SKG mice at 10~12 weeks of age were used for experiments. BALB/c mice were obtained from the Jackson Laboratory (Bar Harbor, MA, USA). All mice were bred and maintained at the animal facility of Seoul National University College of Medicine. All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee at Seoul National University.
Arthritis induction and CP-690550 administration
SKG mice were intraperitoneally injected with curdlan (3 mg for each WAKO, Osaka, Japan). CP-690550 was chemically synthesized, dissolved in polyethylene glycol 300 (Sigma-Aldrich, St Louis, MO, USA), and administered continuously for 14 or 28 days using osmotic mini-pumps (30 mg/kg/day; Alzet, Cupertino, CA, USA). When the clinical score of arthritis reached between 3.5 and 4.0 on average, osmotic pumps containing CP-690,550 were implanted into the recipients. Joint inflammation was monitored by inspection and scored according to the previous report (9) with some modifications: 0.1, swelling of 1 finger joint; 0.5, mild swelling of a wrist or an ankle; 1.0, moderate swelling of a wrist or an ankle; 2.0, severe swelling of a wrist or an ankle. The scores for all digits, wrists, and ankles were summed for each mouse.
Histopathological analysis
Paws from mice placed in 4% buffered paraformaldehyde, decalcified in 5% nitric acid, embedded in paraffin, sectioned, and stained with hematoxylin and eosin for histopathologic analysis.
Intracellular cytokine staining
Peripheral lymph nodes (popliteal and axillary) and the spleen were harvested from mice treated with curdlan, and single-cell suspensions were prepared. Cells were stimulated with 50 ng/ml PMA (Sigma-Aldrich, St Louis, MO, USA) and 1µg/ml ionomycin (Sigma-Aldrich, St Louis, MO) for 4 hours. Brefeldin A (BD-Pharmingen, San Diego, CA, USA) was added for the last 2 hours of incubation. In vitro stimulated cells were stained with antibodies against CD4 and CD8α (BD Pharmingen), fixed with 4% paraformaldehyde (Sigma-Aldrich), and permeabilized with Triton buffer (0.5% Triton X-100 plus 0.1% bovine serum albumin in PBS). Cells were subsequently stained with PE-conjugated anti-IL-17 (TC11-18H10, BD Pharmingen) and APC-conjugated anti-IFN-γ (XMG1.2. eBioscience, San Diego, CA, USA) antibodies. Flow-cytometric analysis was conducted using a FACSCalibur™ system (Becton Dickinson, San Jose, CA, USA), and data were analyzed using Flowjo software (Treestar, Ashland, OR, USA).
RT-PCR
Total RNA was isolated from lung tissues using Trizol (Invitrogen). Complementary DNA generated from 1µg of total RNA by Superscript II (Invitrogen, Carlsbad, CA). The following primer pairs were used for PCR: IL-6, 5'-GACAAAGCCAGAGTCCTTCAGAG-3'/5'-CTAGGTTTGCCGAGTAGATCTC-3'; IL-10, 5'-TGAAGCTTCTATTCTAAGGCTGGCC-3'/5'-CTGAGCTGCTGCAGGAATGATCATC-3'; IL-17, 5'-TCCAGAAGGCCCTCAGACTA-3'/5'-ACACCCACCAGCATCTTCTC-3'; GAPDH, 5'-CCCACTAACATCAAATGGGG-3'/5'-ATCCACAGTCTTCTGGGTGG-3'.
RESULTS
CP-690550 treatment inhibits disease progression of established SKG arthritis
Arthritis was induced in female SKG mice by intraperitoneal injection of fungal protein curdlan (3 mg for each) (28). Joint inflammation became evident from the interphalangeal joints 2 weeks after curdlan injection and was clinically scored by adding the disease scores of all the joints involved according to the previous report (9). To evaluate the therapeutic effect of CP-690550 on established disease, SKG mice with arthritis progression to clinical score between 3.5 and 4.0 were continuously administered with CP-690550 or vehicle using subcutaneously embedded osmotic pumps. CP-690550 treatment halted the disease progression and gradually reduced the severity of arthritis as assessed by clinical score (Fig. 1), whereas in the vehicle-treated control, the severity of arthritis progressed to the maximal clinical score (Fig. 1). Improvement of disease pathogenesis in terms of clinical score continued after cessation of CP-690550 treatment. Therapeutic effect of 14-day treatment of CP-690550 was comparable to 28-day treatment, and the therapeutic effect continued at least 3 more weeks after cessation of CP-690550 infusion (Fig. 1).
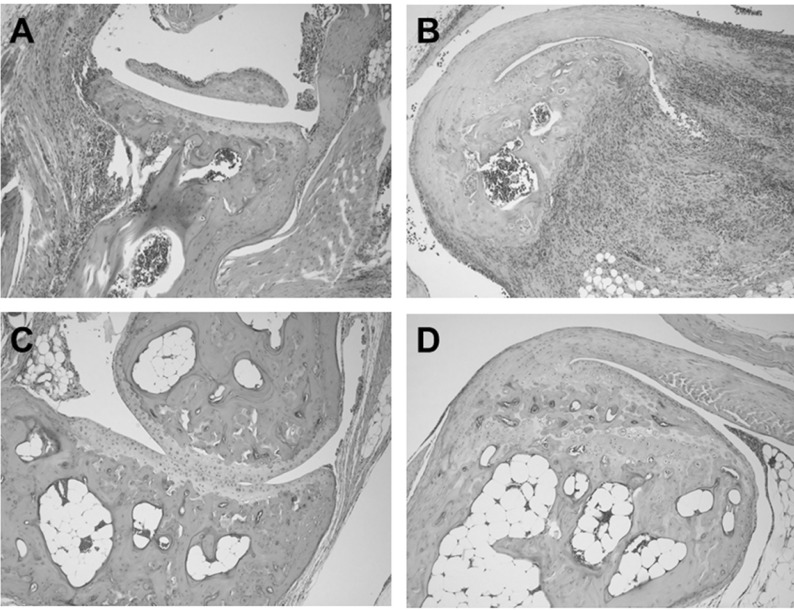
Histopathological analysis of vehicle-treated control mice sacrificed 48 days of after cudlan injection revealed massive inflammatory cell infiltration into the joint tissue and abundant cellular exudates in the joint cavity (Fig. 2A). Mononuclear cells with lymphocyte and macrophage morphology were the predominant infiltrated inflammatory cells, and scattered aggregates of neutrophils were also evident in the articular tissue. Bone erosions were found at several spots in the tibia adjacent to the ankle joint (Fig. 2A). CP-690550 treatment dramatically reduced inflammatory cell infiltration into the joints and most of the remaining cells were mononuclear cells with little neutrophil infiltration (Fig. 2B). Bone erosion due to pathogenesis of the disease was not visible in the CP-690550-treated group at this time point (Fig. 2B).
CP-690550 treatment inhibited Th17 differentiation of CD4+ T cells
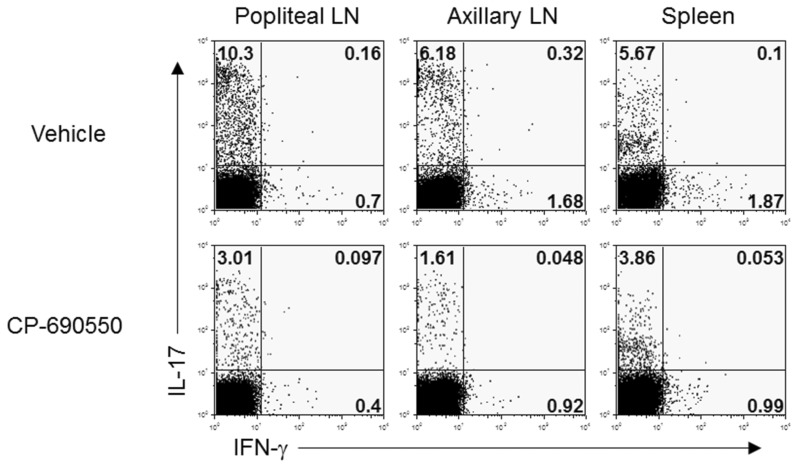
To evaluate underlying immunopathological differences following CP-690550 treatment, lymphocytes from draining popliteal and axillary lymph nodes and splenocytes were obtained on day 18 and stimulated ex vivo with PMA and ionomycin for 4 h, and intracellular cytokine analysis was performed. In agreement with the previous reports, IL-17-producing T-helper (Th17) cells were predominant in the curdlan-injected arthritic SKG mice, with a few IFN-γ-producing T-helper cells in secondary lymphoid organs (Fig. 3). CD4+ IL-17+ cells in lymphoid organs were dramatically decreased by CP-690550 treatment compared to the vehicle-treated control mice (Fig. 3). Quantitative RT-PCR analysis of the affected joint tissue on day 18 revealed increased proinflammatory cytokines, including IL-6, IL-17, and IFN-γ in the vehicle-treated control mice, and all of the cytokines were reduced in the articular tissue of the CP-690550-treated SKG mice (Fig. 4). Major immunoregulatory cytokine, IL-10 was increased in the CP-690550-treated mice compared to the vehicle-treated control mice (Fig. 4).
DISCUSSION
SKG mice are originally characterized by spontaneously developing arthritis. An altered codon 163 from tryptophan to cysteine (W163C) through point mutation of zeta-chain-associated protein kinase 70 (ZAP-70), the critical signaling molecule of T-cell receptors (TCRs), is the key genetic defect in SKG mice (27). Abnormal ZAP-70 molecules in SKG mice thus lead to decrease in the signaling strength of TCRs and peptide-MHC antigen interactions. Consequently, appropriate signaling adjustments during the positive and negative selection of autoreactive T cells in the thymus are compromised (29), and the mature T cells of SKG mice show inherent autoreactivity to self antigens (27,29). SKG mice spontaneously develop chronic arthritis, which is characterized by subsynovial inflammatory cell infiltration, proliferation of synoviocytes, pannus formation, and accompanying neovascularization, which closely resembles articular manifestations of human RA (27). Moreover, SKG mice also reveal systemic manifestations characteristic of human RA, such as interstitial pneumonitis and subcutaneous nodules (1,4,27). As SKG mice show similar immunological, histopathological, and clinical features as human RA, evaluation of long-term therapeutic effects of CP-690550 on established arthritis in an SKG model is important in translating therapeutic effect of this newly developed JAK inhibitor to human RA.
CP-690550 was originally developed through in vitro kinase inhibition assay (17), and the sensitivity and specificity of the drug was also confirmed using cell-based co-transfection assay in our previous report (30). In this study, we found that administration of CP-690550 following disease progression to intermediate-to-severe joint disease dramatically reduced the pathogenesis of established chronic arthritis in an SKG mice model, and suppression of joint inflammation was correlated with reduced inflammatory cell infiltration and proinflammatory cytokine expressions including IL-6, IL-17, and IFN-γ. Moreover, significantly increased levels of IL-10, a major immunoregulatory cytokine (31,32), in the CP-690550-treated mice compared to the vehicle-treated control mice explained the long-term therapeutic effects of CP-690550. In addition, the key inhibitory effect of CP-690550 include the decrease in arthritogenic Th17 cell differentiation, which, can directly damage joint tissues and provoke a vicious inflammatory cascade recruiting more inflammatory cells, such as neutrophils, into the battleground by producing IL-17 (13,16). The major pathogenic T-helper subset in SKG arthritis was Th17, and effective inhibition of Th17 differentiation by CP-690550 in this study was intriguing in that suppression of Th1 cells with relative sparing of Th17 differentiation was our critical findings of the allogeneic graft versus host disease (GVHD) model treated with CP-690550 in our previous report, where the predominant pathogenic effector helper-T subset was Th1 (21). Thus CP-690550, through inhibition of multiple cytokine signaling, efficiently controlled the major pathogenic T-helper subsets in different disease models depending on the context of specific immunological derangements.
Treatment of established autoimmune diseases needs to deal with activated/memory T cells which are more refractory to the treatment compared to naïve unactivated T cells (33,34). The effectiveness of a wide variety of immunomodulating reagents has been well documented in which these reagents have been used at or during the disease induction phase. However, many reagents failed to work when they were administered to the well-advanced disease where expanded activated T cells prevailed (35). In this respect, the dramatic effect of CP-690550 on established arthritis is very impressive and quite promising in that most of the pathogenic autoreactive T cells are already activated at the time of diagnosis in human autoimmune diseases (2,5,33).
In this study, the long-term therapeutic effect of CP-690550 even after cessation of drug infusion supports the long-lasting treatment effect of CP-690550 in an acute allogeneic GVHD model of our previous report, whose effect lasted over 60 days following administration of CP-690550 during -2 to 12 days of disease induction (21). At that time, we assumed that compromised receptor signaling by CP-690550 during naïve T-cell activation could cause tolerance induction of allogeneic T cells which leads to long-term unresponsiveness of these cells to cognate antigens. In this study, however, we targeted fully activated pathogenic T cells and still obtained very dramatic long-lasting effects. We are now investigating molecular and cellular mechanisms underlying these unexpected long-term therapeutic effects.


 XML Download
XML Download