

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Under homeostatic conditions, bone mass and architecture is maintained by a balance of activity between the effects of osteoblasts to form bone and those of osteoclasts to resorb bone. It has long been appreciated that chronic inflammation disrupts this balance, producing both local and systemic bone loss. Local erosions of bone surrounding inflamed joints are characteristic of rheumatoid arthritis. Inflammation in inflammatory bowel disease and a number of other autoimmune disorders produces systemic osteopenia and entails a risk of bone fractures (1). In some cases, bone loss accompanied by specific autoimmune disorders is remarkably severe, such as the profound loss of bone seen in the autoimmune disorder Cherubism (2). Naturally emerging from these clinical observations have been seminal observations describing mechanisms by which immune cells and inflammatory cytokines mediate bone catabolism. However, beyond these findings, a more textured understanding of interactions between the immune and skeletal systems is emerging, and recent data demonstrate that the immune system may additionally play pro-anabolic functions. Furthermore, the role of the skeleton in fostering the development of immune lineages has received increasing appreciation. Collectively, these and similar observations regarding cross-regulation between the immune and skeletal systems constitute the field of osteoimmunology. Here we briefly highlight core areas of interest and selected recent advances in this field.
Go to : 
THE EFFECT OF THE IMMUNE SYSTEM ON THE PROMOTION OF BONE CATABOLISM
Classic observations regarding osteoimmunology establish the effects of pro-inflammatory cytokines, such as TNF, on the promotion of bone catabolism. In the context of inflammatory arthritis, the best studied mediator is TNF, which is both necessary and sufficient to trigger bone breakdown. TNF acts via at least 3 distinct mechanisms (3). First, TNF inhibits the differentiation and bone-forming activity of osteoblasts. Additionally, TNF promotes stromal cells to express receptor activator of NF-κB ligand (RANKL), a key cytokine that acts via its receptor RANK on osteoclast precursors to drive osteoclast differentiation. Lastly, TNF acts directly on osteoclasts to potentate their differentiation and increase their resorptive capacity. Accordingly, treatment with TNF-blocking biologics increases bone mass in many inflammatory disorders, including rheumatoid arthritis, juvenile rheumatoid arthritis, ankylosing spondylitis, and Crohn's disease, though in some cases it may be difficult to deconvolute the specific effects on bone versus overall effects of disease remission (4). IL-6 and IL-1 also appear to have similar, albeit less well characterized effects to promote bone resorption.
Notably, in addition to these, IL-17 and Th17 cells have been identified as playing a potent and specific role in promoting bone resorption (5,6). This effect is dependent on IL-17, which acts on stromal cells and osteoblasts to drive up-regulation of RANKL and promote osteoclast differentiation. In contrast, the role of Th1 and Th2 cells in osteoclast differentiation appears to be less straightforward and highly context dependent. In vitro, Th1 and Th2-skewed T cells inhibit osteoclast development via their characteristic cytokines IFN-γ and IL-4, respectively (6). In particular, IFN-γ effects inhibition of osteoclast differentiation by enforcing downregulation of TRAF6, a crucial mediator connecting RANK activation to activation of the NF-κB family of transcription factors, that in turn promote osteoclast differentiation (7). In vivo, however, IFN-γ has an additional indirect effect on the expressions of TNF and RANKL, and under the conditions of estrogen withdrawal these pro-catabolic effects predominate over the direct effect of IFN-γ on the suppression of osteoclast differentiation (8) (Fig. 1).
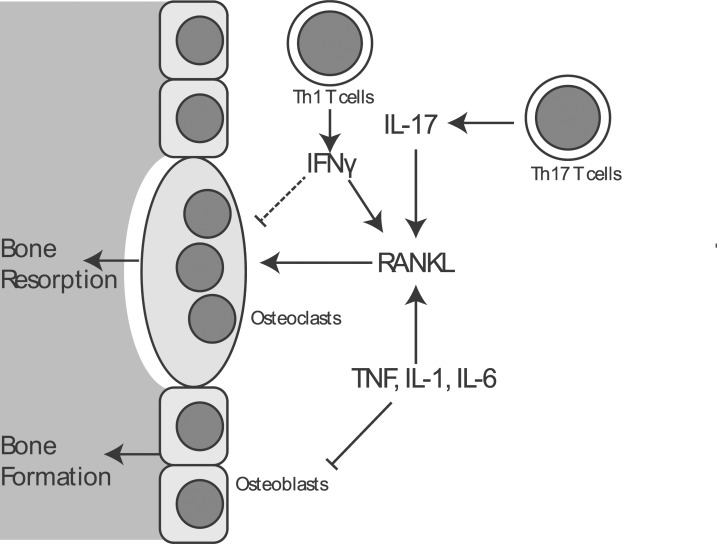
Likewise, in a model of bone formation by subcutaneous implantation of mesenchymal progenitors of osteoblasts, the basal presence of T cells inhibits bone formation by these implanted cells (9). This inhibition of bone formation mapped to production of both IFN-γ and TNF. IFN-γ blocked osteoblast differentiation by inhibiting induction of RUNX2, a transcription factor that functions as a master regulator of osteoblast differentiation. Additionally, IFN-γ synergizes with TNF to induce apoptosis of the implanted mesenchymal progenitors. Given that implantation of mesenchymal progenitors may ultimately be an attractive approach for the management of skeletal fractures, these findings highlight that preventing immune-mediated suppression of bone formation will be an important hurdle such therapies will need to overcome to be successful. T cells also appear to interact with the hormone axes that control bone mass. In a model of hyperparathyroidism generated by continuous infusion of parathyroid hormone (PTH), the presence of T cells promotes the ability of PTH to mediate bone loss (10), as T cells mediate osteoclast generation in this context via CD40L expression. CD40L in turn stimulates stromal cells to express RANKL, and additionally, CD40L stimulation suppresses expression of osteoprotegrin (OPG), a secreted decoy receptor for RANKL that down-regulates the activity of the key RANKL/RANK axis to promote osteoclast differentiation.
Lastly, immune cells have also been of great interest as a potential source of RANKL, as this would allow for direct induction of osteoclast differentiation. Since RANKL is expressed by T cells, it was initially proposed as the major mechanism by which T cells mediate pro-catabolic effects on bone. As mentioned above, however, the anti-osteoclastogenic effects of IL-4 have made demonstration of a direct effect of Th1 or Th2 cells on the promotion of osteoclast differentiation difficult. Notably, B cells also express RANKL, while a T cell-specific knockout of RANKL displays no protective effects on bone mass in a mouse model of sex hormone withdrawal-induced osteoporosis following ovariectomy, specific deletion of RANKL in B cells blunts the bone loss and increases the number of osteoclasts, which are the characteristics of this mouse model. Thus, in addition to the ability of lymphocytes to induce RANKL expression on stromal cells, B cells themselves serve as a physiologically relevant source of RANKL and are potentially relevant to the physiology of post-menopausal osteoporosis.
Go to :
THE EFFECT OF THE IMMUNE SYSTEM ON THE PROMOTION OF BONE ANABOLISM
Over the last few years, surprising new evidence has complicated the straightforward model of immune activation-producing bone catabolism, suggesting a more complex model that certain immune response pathways additionally promote bone formation. In addition to the effects of the adaptive immune system on bone mass described above, innate immune pathways also regulate bone mass. Injection of LPS or bacterial lipoproteins stimulates osteoclast differentiation in vitro (11). Injection of high concentrations of LPS into the calvarium stimulates local bone resorption in vivo. Interestingly, while mice lacking MyD88-which is a crucial adaptor protein required for many aspects of the response to LPS and other selected microbe-associated molecules-display a reduced number of osteoclasts and their overall bone mass is reduced. Bone formation and bone resorption are often coupled, with a decrease in bone resorption triggering a commensurate decrease in bone formation. Thus, the defect in osteoclasts in MyD88-deficient mice may in turn cause a low rate of bone formation, resulting in low bone turnover osteopenia. These findings suggest that a relatively low level of immune stimulation is necessary to maintain the basal bone turnover in order to avoid adynamic bone disease.
One outstanding question in this respect is what is the source of stimulatory ligands that activates MyD88-dependent pathways. One possibility is that the ligands are derived from gut microbiota; however, germ-free mice have recently been shown to have an increase in bone mass (12). Thus, MyD88 is not a dominant pathway affecting bone engaged by gut flora, or alternatively MyD88 activating-signals may not be microbially derived, but instead may come from endogenous "danger signals" liberated during normal cellular turnover. Additionally, other cytokines, such as IL-1 and IL-18, also signal through MyD88, and defective signaling by these cytokines may also contribute to the phenotype of MyD88-deficient mice.
In addition to serving as a source of cytokines that modulate bone formation, T cells are also a source of growth factors that modulate bone formation. In contrast to the bone loss induced by hyperparathoidism, treatment with exogenous PTH induces bone formation due to differences in the kinetics of release, and this is a widely utilized approach to stimulate bone formation in the setting of osteoporosis. Surprisingly, despite well documented direct effects on osteoblasts, the effects of anabolic PTH depend on the ability of T cells to secrete the growth factor Wnt10b (13). Thus, T cells appear to be involved in mediating both anabolic and catabolic effects of PTH, each through distinct pathways.
Lastly, insofar as pro-inflammatory pathways can promote bone resorption, immunoregulatory pathways can promote maintenance of bone mass. However, beyond nonspecific effects to control inflammation, regulatory T cells can directly inhibit osteoclastogenesis. Co-culture with human regulatory T cells inhibits osteoclast differentiation in vitro in a IL4-and TGFβ-dependent manner (14). Additionally, the negative regulatory costimulatory molecule expressed on Tregs, cytotoxic T lymphocyte antigen 4 (CTLA4), has also been shown to mediate the inhibition of osteoclast differentiation (15). Accordingly, in a model of arthritis through transgenic expression of TNF, which will be insensitive to confounding effects of Tregs on primary inflammatory stimulus, Tregs exerts a protective effect in terms of both local and systemic bone loss (16). Similarly, Foxp3 transgenic mice with an increased number of Tregs display increased basal bone mass and partial protection against bone loss induced by sex hormone withdrawal after ovariectomy (17). Adoptive transference of Tregs to Rag-1-/- mice otherwise lacking T and B lymphocytes also increases basal bone mass. Thus, immunomodulatory mechanisms not only appear to play a role in modulating bone turnover in the context of specific inflammatory or autoimmune disorders, but they also appear to contribute to the maintenance of bone mass even in the absence of an overt inflammatory stimulus.
Go to :
BONE AS A CRADLE FOR IMMUNE SYSTEM DEVELOPMENT
While it has long been appreciated as a matter of fact that bone houses immature hematopoetic stem cells (HSCs), it has only recently been appreciated that osteoblasts and other bone stromal cells contribute to the specific molecular environment necessary for HSC maintenance, which has been termed the "HSC niche". Key studies have focused on the role of expression of the Notch ligand jagged-1 by osteoblasts in an increasing number of osteoblasts. Expression of a constitutively active PTH/PTH-related peptide receptor in osteoblasts increases the number of osteoblast numbers and jagged-1 expression and the total number of HSCs (18). As activation of the PTH axis is a current therapeutic approach for the treatment of osteoporosis, these results suggest that the concurrent use of PTH may have benefits in improving HSC engraftment after bone marrow transplantation. Additionally, most HSCs are maintained in a non-cycling G0 state, and this basal quiescence is an important feature for sustaining hematopoietic functions of HSCs. Angiopoietin-1 secreted by osteoblasts has emerged as an important molecular determinant of HSC quiescence, stimulating the angiopiopoietin-1 receptor Tie-2 on HSCs to promote HSC adherence within bone marrow (19). Not only can bone influence hematopoesis, but under selected conditions, disorders of hematopoiesis can also influence bone mass (20). In thalassemia, iron homeostasis is perturbed, leading to inappropriate retention of iron and eventual iron overload. The excess iron that accrues has a negative impact on bone physiology, leading to high bone turnover osteopenia. Interestingly, these changes are correlated with increases in proinflammatory cytokines and could be partially reversed by treatment with the antioxidant N-acetyl cystine, arguing that the redox imbalance induced by iron may act via inflammatory mediators to affect bone mass.
Despite these advances, however, it still remains unresolved to what degree HSC dynamics and properties of the HSC niche are generally coupled with bone metabolism. Is bone health a factor that should be evaluated and optimized prior to HSC transplantation? While disorders of bone metabolism occluding the marrow space are well appreciated to disrupt hematopoiesis, can other, more subtle disorders of bone metabolism also affect hematopoiesis? Answers to these and similar questions await extension of studies of the HSC niche into further models of bone metabolic disorders.
Go to :
SUMMARY AND FUTURE DIRECTIONS
Accruing evidence over approximately the past decade supports a model that interactions between the immune system and bone extend far beyond T cells simply promoting local bone erosion in the context of inflammatory arthritis. Additional data supports that both innate and adaptive arms of the immune system contribute to basal bone homeostasis, even outside the context of specific inflammatory disorders. Additionally, interactions between bone and the immune system are bidirectional, insofar as bone is a home for HSCs and the early stages of differentiation for both myeloid cells and lymphocytes.
Many potential lines of future investigation are open at this relatively early stage for the field of osteoimmunology. So far, investigation of the interactions between immune cells and bone have mostly focused on interactions between osteoclasts and immune cells, with a smaller amount of additional work characterizing the effects on osteoblasts. However, how the immune system may interact with other cell types comprising bone, growth plate chondrocytes, and osteocytes, remains unclear. Additionally, many arms of the innate immune system remain uncharacterized regarding their effects on bone, as the contributions of granulocytes, macrophages, or non Toll-like receptor (TLR) innate immune pathways to basal bone homeostasis remain poorly characterized. Lastly, it is also mostly unknown how bone homeostasis may in turn shape the immune system. One suggestion of such an effect comes from longstanding observations that, while not displaying a strong correlation with the extent of disease, serum erythrocyte sedimentation rates (ESR), a classic marker of inflammation, tend to be higher in patients with Paget's disease of bone, a disorder characterized by extraordinarily high rates of bone turnover (21). While further work needs to be done to exclude that this is an unrelated epiphenomenon, it does suggest the possibility that bone turnover may influence inflammation.
Additionally, while the insights of osteimmunology have obvious relevance to the pathogenesis of bone erosions seen in rheumatoid arthritis and similar disorders, many of the recent studies discussed above demonstrate that lymphocytes are involved in growth factor production, hormone responses, and the pathophysiology of bone loss induced by withdrawal of sex hormones. These findings prove that osteoimmunology is a field with broad and general relevance to bone metabolism. Undoubtedly, the next decade of osteoimmunology research will continue to expand that relevance and shed light on the deeply intertwined nature of bone metabolism and immune function.
Go to :
 XML Download
XML Download