

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abbreviations
LC3
microtubule-associated protein1 light chain 3
ESX-1
spermatogenic homeobox 1
BCG
Bacille Calmette-Guérin
PAMPs
pathogen-associated molecular patterns
PRRs
pattern recognition receptors
TLRs
toll-like receptors
NLRs
Nucleotide oligomerization domain (NOD)-like receptors
RLRs
Retinoic acid-inducible gene (RIG)-I-like receptors
MDA-5
Melanoma differentiation associated gene 5
INTRODUCTION
Autophagy, ancient system necessary to maintain homeostasis in eukaryotic cells, degrades long-lived cytoplasmic proteins and organelles and provides nutrients in starvation or stress conditions (1). There are at least three types of autophagy in eukaryotic cells: macroautophagy, microautophagy, and chaperone-mediated autophagy (2). Microautophagy involves the continuous degradation of cytosolic constituents close to the lysosomes via budding of the lysosomal membrane. In chaperone-mediated autophagy, proteins containing a KFERQ motif, which is recognized by Hsc70, are transported into the lysosomal lumen via Lamp-2a to be degraded (3-6). Macroautophagy is the primary route of degradation and has been most widely studied. In this process, cellular components such as protein aggregates and organelles (e.g., mitochondria) are wrapped by an elongated cup-shaped membrane, which forms a double-membrane structure called an autophagosome (7). Autophagosomes mature and fuse with lysosomes to facilitate the degradation of the internal contents. Recently, increasing evidence suggests that autophagy pathway involves selective process. In selective autophagy, autophagic adaptors such as p62 targets polyubiquitylated substrates and are selectively degraded by autophagy (8).
The autophagic pathway consists of three distinct stages. Initially, an isolation membrane is formed through complex interaction of the autophagic protein Atg6 (Beclin-1) and type-III phosphatidylinositol 3-kinase, along with membrane components. Elongation of the isolation membrane and completion of autophagosome formation are regulated by at least two ubiquitin-like systems: the microtubule-associated protein 1 light chain 3 (LC3; mammalian homolog of the yeast autophagic protein Atg8) and the Atg12 conjugation systems (9,10). Atg12 is transferred to the E1- and E2-like enzyme Atg7 and Atg10 sequentially, and is finally conjugated with a lysine residue of Atg5 through its C-terminal glycine residue. The Atg12-Atg5 conjugates facilitate the elongation of the isolation membrane and catalyze the LC3 conjugation with the help of Atg16. The C-terminal amino acids of LC3 are cleaved by Atg4 and conjugated with phosphatidylethanolamine (PE) by the E1- and E2-like enzymes Atg7 and Atg3, respectively. The lipidated LC3 (LC3-II) is incorporated into the newly formed autophagosomal membrane. Upon autophagosome closure, the Atg12-Atg5-Atg16 complex dissociates from the outer autophagosomal membrane, but LC3 remains in the autophagosomal lumen as an autophagosomal marker. Interestingly, LC3 binds to the adaptor protein p62, which interacts with polyubiquitylated structures to facilitate their autophagic degradation (11). Finally, the outer membrane of the autophagosome fuses with the lysosomal membrane, followed by degradation of the membrane and its contents.
Although autophagy is necessary for cellular homeostasis, it is involved in biological processes including development, aging, and degeneration (12). Aberrant regulation of autophagy is related to many diseases, such as cancer and neurodegenerative disease (13,14). Furthermore, recent studies have identified the important roles of autophagy in immunity. Autophagy can directly eliminate intracellular pathogens or enhance pathogen recognition and elimination (15-19), and can regulate autoimmunity and inflammatory disorders (20). Moreover, autophagy affects the adaptive immune system by contributing to antigen presentation and controlling T- and B-cell homeostasis (21-26). Here, we briefly review the role of autophagy in innate immune control.
DIRECT ELIMINATION OF PATHOGENS BY AUTOPHAGY
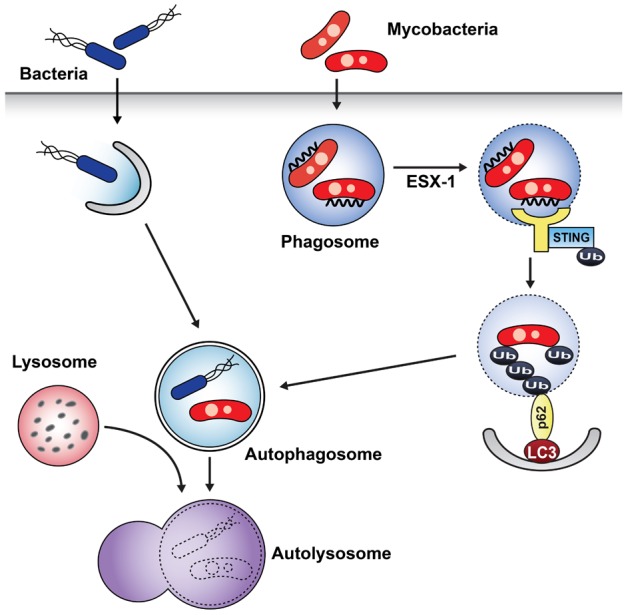
Because autophagy removes large cytoplasmic proteins and organelles, intracellular pathogens such as bacteria and parasites are cleared by autophagic machinery. Autophagy can target free microbes in the cytosol and pathogens contained in phagosomes (27). A study by Nakagawa et al. (15) showed that group A Streptococcus (GAS) that escapes from endosomes into the cytoplasm becomes enveloped by autophagosome-like compartments and is killed upon fusion of these compartments with lysosomes (Fig. 1). GAS survives and multiplies in Atg5-deficient cells, which indicates that elimination of GAS is autophagy-dependent.
In cases of intracellular pathogens that persist within phagosomes such as Mycobacterium tuberculosis, autophagy promotes the maturation of mycobacterial phagosomes into phagolysosomes. Using Mycobacterium bovis Bacille Calmette-Guérin (BCG), starvation- or rapamycin-induced autophagy leads to mycobacterial phagosome colocalization with the LC3, resulting in fusion of phagosomes with autophagosomes, which in turn deliver the pathogen-containing vacuoles for lysosomal degradation (16). In case of human macrophages, vitamin D treatment stimulates autophagy activation against M. tuberculosis through induction of cathelicidins. It induces promoter activation of the autophagy-related genes Atg5 and Beclin-1, and promotes colocalization of bacterial phagosomes and autophagosomes (28).
However, the mechanism of autophagy targeting and its role in natural infection without exogenous induction of autophagy remains unclear. Interestingly, a recent study using wild type Mycobacterium tuberculosis revealed how M. tuberculosis interfaces with the selective autophagy pathway from within the phagosomes in resting macrophages (Fig. 1) (29). Unlike BCG, the attenuated vaccine strain, M. tuberculosis includes several virulence factors such as the extraembryonic spermatogenic homeobox 1 (ESX-1) secretion system (30, 31). The bacterial ESX-1 facilitates the exposure of bacterial DNA to the host by permeabilizing the phagosome membrane (32). The exposed bacterial DNA is recognized by the cytosolic DNA pathway, including stimulator of interferon (IFN) genes (STING), and surrounded by a ubiquitin chain. Ubiquitin and LC3-binding autophagy adaptors p62 and nuclear dot protein 52 (NDP52) recruit autophagy components to target the bacilli to the selective autophagy pathway. In this process, Atg5 and tank-binding kinase 1 are also required. Consequently, bacilli-containing autophagosomes are fused with lysosomes to facilitate the elimination of mycobacteria. Other intracellular bacteria and parasites such as Listeria monocytogenes and Salmonella species are also limited by autophagy with various strategies including selective autophagy activation (33-36).
INTERACTION BETWEEN AUTOPHAGY AND TOLL-LIKE RECEPTORS
The innate immune system recognizes conserved microbial molecular structures, so called pathogen-associated molecular patterns (PAMPs). Pattern recognition receptors (PRRs) bind to these conserved structures and initiate downstream signaling pathways (37). In addition, signaling initiated by PRR activation can promote the autophagy induction. Studies have shown that activation of Toll-like receptors (TLRs) facilitates pathogen elimination by autophagy induction (17, 18). TLR4 stimulated with lipopolysaccharide (LPS) induces autophagy in primary human macrophages and the murine macrophage cell line, RAW 264.7. Redistribution of LC3 protein from a diffuse to a punctate pattern and increased levels of the lipidated form of LC3 (LC3II), both of which are reliable markers of autophagy induction, were observed after stimulation with LPS. This process occurred via the toll/interleukin-1 receptor domain-containing adapter-inducing interferon β (TRIF)-p38 axis, but not via MyD88, and resulted in formation of the autophagosome colocalized with mycobacteria (Fig. 2A) (17). Thus, it was suggested that autophagy induced by TLR activation enhances the elimination of mycobacteria.
According to a report showing the effect of TLR agonists on autophagy induction in RAW264.7 (18), ligands of TLR3, TLR4, and TLR7 could induce autophagy and ligands of TLR7 generate the most potent effects. TLR7 signaling activated by two different ligands, single-stranded RNA (ssRNA) and imiquimod, induces the formation of autophagosomes characterized by LC3 puncta formation in murine macrophages (Fig. 2A). This process is dependent on MyD88 and requires Beclin-1. TLR7-induced autophagy activation promotes the killing of intracellular mycobacteria, even though mycobacteria are normally not associated with TLR signaling (18).
In addition to the formation of autophagosomes fused with pathogen-containing phagosomes, TLR signaling could enhance the maturation of phagosomes into phagolysosomes via autophagic machinery (19). When zymosan (a component of the fungal cell wall) is phagocytosed, phagosomes rapidly recruit LC3 and fuse with lysosomes (Fig. 2B). Pam3CSK4-coated latex beads also induce rapid recruitment of LC3 to phasogomes in RAW264.7. LC3 translocation to phagosomes was dependent on TLR2 but not MyD88, and requires Atg 5 and Atg7. Interestingly, LC3 recruitment to phagosomal membranes is not associated with the double-membrane structures characteristic of autophagosomes. Thus, this study identified a new way of using the autophagic machinery to enhance conventional functions of phagocytes after TLR activation (38).
AUTOPHAGY IN NUCLEOTIDE OLIGOMERIZATION DOMAIN-LIKE RECEPTOR SIGNALING
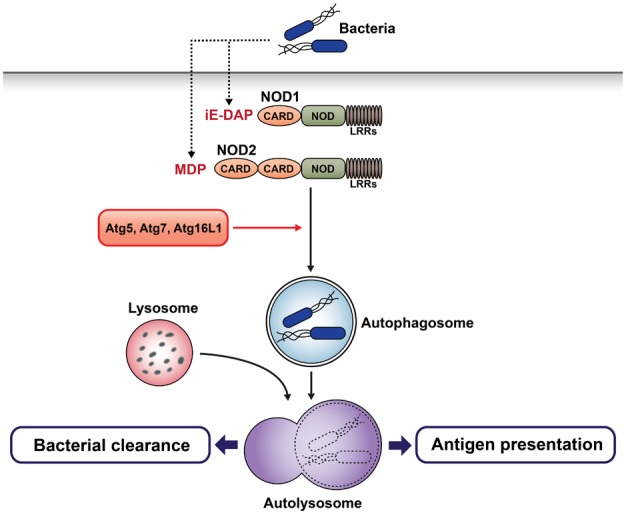
Nucleotide oligomerization domain (NOD)-like receptors (NLRs) are a family of cytoplasmic molecules that recognize bacterial cell wall components, such as peptidoglycan, in the cytosol. Upon bacterial invasion, NLRs recognize bacterial peptidoglycan by their C-terminal leucine-rich repeat and initiate signal transduction by their N-terminal effector domain. Downstream signals activate nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and activator protein 1, and lead to the production of cytokines involved in innate defense (39). Studies have shown that NOD1 and NOD2 are critical for the autophagic response to bacterial invasion (40, 41). In mouse embryonic fibroblasts (MEFs), NOD2 recruits Atg16L1 to the plasma membrane at the bacterial entry site, which facilitates bacterial trafficking to the autophagosomes. This process does not require the adaptor receptor interacting protein 2 and the transcription factor NF-κB (41). Another study showed that NOD2 activation by muramyl dipeptide (MDP) induces autophagosome formation and subsequently promotes major histocompatibility complex (MHC) class II-associated antigen presentation in human dendritic cells (DCs). In this process, Atg5, Atg7, Atg16L1, and receptor-interacting serine-threonine kinase 2 are required (40). Thus, autophagy mediates bacterial clearance and bacterial antigen presentation, which are initiated by NOD sensing of invasive bacteria (Fig. 3). Interestingly, NOD2 and Atg16L1 are two of the most important genes associated with Crohn's disease (42-44), a complicated inflammatory condition. DCs isolated from patients with Crohn's disease expressing risk alleles for NOD2 and Atg16L1 are impaired in autophagy induction, bacterial trafficking, and antigen presentation. These reports reveal the functional relationship between NOD2 and Atg16L1 in Crohn's disease.
AUTOPHAGY IN ANTIVIRAL IMMUNITY
Autophagy plays a role in directly controlling viral pathogenesis. The first-known study is about Sindbis viral infection. Overexpression or neuron-specific deletion of ATG proteins resulted in increased or decreased survival following intracranial injection with Sindbis virus, respectively (45,46). In another study, intracranial infection with HSV-1 beclin-1 binding deficient mutant resulted in improved survival and decreased HSV-1 replication (47). Virulence factor, ICP 34.5, encoded by herpes simplex virus 1 (HSV-1) blocks autophagy induction through binding and inhibiting beclin-1. Thus, these results showed that autophagy controls HSV-1 replication (48).
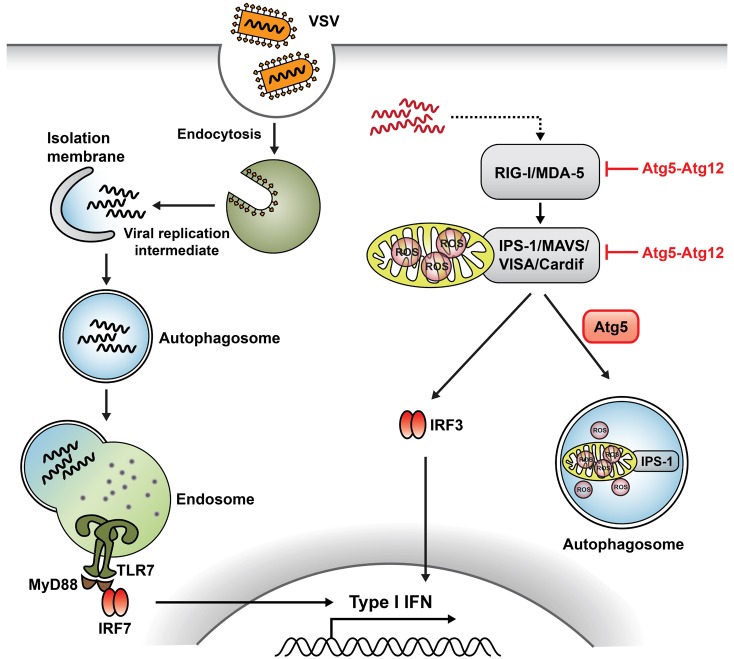
Autophagy also supports antiviral defense by delivering cytosolic viral PAMPs to endosomal TLRs. Most viral sensing is mediated by viral nucleic acids, which is different from other pathogens containing particular cell wall components triggering TLR activation. TLR3, TLR7, TLR8, and TLR9 sense endocytosed viral nucleic acids in the endosomal compartment (49). In plasmacytoid DCs (pDCs), viral RNA or DNA recognized by TLR7 or TLR9, respectively, induces IFN regulatory factor 7 (IRF7) activation via MyD88, leading to type-I IFN production. In vesicular stomatitis virus (VSV), pDCs recognize replicating virus in cytosol rather than the viral genome via TLR7 in endosomes. However, how these cytosolic replication intermediates gain entry to the endosomal compartment where TLR7 resides is not completely known. A study demonstrated that autophagy mediates the delivery of the cytosolic PAMP to the lysosomes to activate TLR7 signaling (Fig. 4) (50). Thus, Atg5-deficient pDCs are unable to produce IFN-α and interleukin (IL)-12p40 in response to VSV. Consequently, mice lacking Atg5 are susceptible to systemic VSV infection in vivo. In addition to VSV, pDCs lacking Atg5 failed to produce IFN-α in response to HSV-1, which is recognized by TLR9, whereas the IL-12 response was not impaired in these cells. Autophagy plays an important role in the antiviral response by delivering viral replication intermediates from the cytosol to the endosome. However, the precise mechanism of the differential control of NF-κB versus IFN-α induction pathways in pDCs by autophagy remains to be determined (50-52).
Autophagy-related proteins also regulate the antiviral immune response in another pathway. Most cell types other than pDCs use cytosolic receptors, retinoic acid-inducible gene (RIG)-I-like receptors (RLRs) and melanoma differentiation associated gene 5 (MDA-5), to sense the viral invasion (53-55). Via their caspase-recruiting domains (CARDs), RLRs recognize double-stranded RNA, which is synthesized during active viral replication in the cytosol and signal through IFN-β promoter stimulator-1 (IPS-1; also known as MAVS, VISA, or Cardif), and activate transcription factors IRF-3 and NF-κB, subsequently leading to the production of type-I IFN and proinflammatory cytokines (39). A study showed that Atg5-Atg12 conjugates negatively regulate innate viral recognition by RIG-I and MDA-5 in MEFs (56). The Atg5-Atg12 conjugates, which are directly associated with CARD domains of RIG-I and IPS-1, block RLR signaling and suppress type-I IFN production (Fig. 4). Consequently, Atg5- and Atg7-deficient MEFs, lacking Atg5-Atg12 conjugates, overproduce type-I IFN in response to VSV. Similarly, Atg5-deficient cells show increased IFN production through enhanced RLR signaling in response to VSV infection. In this study, Atg5-deficient cells accumulate reactive oxygen species (ROS) localized to the mitochondria. In addition, dysfunctional mitochondria and mitochondria-associated IPS-1 are accumulated in the absence of autophagy. Collectively, accumulation of ROS associated with dysfunctional mitochondria may potentiate RLR signaling in Atg5-deficient cells (Fig. 4) (57). Thus, autophagy acts as an important modulator for balancing the innate antiviral response.
AUTOPHAGY IN REGULATION OF INFLAMMASOME
Inflammasomes are molecular machinery that promote innate immune defenses by triggering the maturation of proinflammatory cytokines such as IL-1β (58). Recent studies reported the complex interplay between inflammasomes and autophagy (19,52). Stimulation of TLR4 by LPS is unable to induce inflammasome activation in macrophages if LPS is not contaminated with other ligands. However, Atg16L1-deficient macrophages exhibit enhanced IL-1β and IL-18 production in response to LPS (Fig. 5A), suggesting that autophagy normally suppresses inflammasome activation by LPS (20). Atg16L1 is an essential component of the autophagosome, forming a complex with Atg5-Atg12 conjugates and inducing LC3-PE conjugation. Interestingly, Atg16L1 is one of the most important genes associated with Crohn's disease. Thus, endotoxin-induced inflammasome activation in Atg16L1 deficiency could be involved in the occurrence of Crohn's disease.
In a recent study detailing the complex reciprocal regulation of inflammasome and autophagic pathways, blocking autophagy potentiated the inflammasome activity, whereas stimulating autophagy limited it (59). The activation of absent in melanoma 2 (AIM2) or NLR pyrin domain containing 3 (NLRP3) inflammasomes trigger autophagy induction by activating RalB to bind to Exo84, which serves as platform for the formation of isolation membrane (60). Autophagy engulfs ubiquitylated assembled inflammasomes through p62, an autophagic adaptor including both ubiquitin-associated domains and LC3-interacting regions, that recognizes ubiquitinated molecules and assists their degradation by autophagy (Fig. 5B) (8,59). Thus, activation of inflammasomes induces autophagy, which in turn limits the inflammasome activity by physical engulfment. This may represent a negative regulation of autophagy in maintaining homeostasis by returning to a basal state in some inflammatory conditions.
CONCLUSION
Recent advances in the characterization of autophagic machinery enable us to identify the role of autophagy in immune systems. By virtue of its ability to degrade cytosolic constituents, autophagy has been shown to eliminate intracellular pathogens regardless of their location (cytosol or phagosome). Moreover, autophagic machinery induced by TLR activation facilitates clearance of pathogens through autophagosome formation or maturation of phagosomes. Autophagy also mediates bacterial clearance and adaptive immune responses including antigen presentation to MHC class II after bacterial sensing via NLRs. In viral infection, autophagy promotes the production of antiviral type-I IFN by delivering the cytosolic replication intermediates to the lysosomes, enabling recognition of virus by endosomal TLRs. Autophagy may also play a critical role in regulating inflammatory conditions including Crohn's disease by controlling inflammasome activity. Autophagy prevents excessive activation of inflammasomes to maintain basal status against endogenous/exogenous irritants. Collectively, autophagy plays multiple roles in immunity, either activating or suppressing immune responses. Comprehensive understanding of autophagy will therefore provide a more integrated picture of how this system controls immune responses.


 XML Download
XML Download