

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Encephalitis is "the presence of an inflammatory process of the brain associated with clinical evidence of neurological dysfunction," as defined by the Infectious Diseases Society of America (1). Though more than one hundred different infectious agents that cause acute encephalitis have been identified, viral infections are responsible for the majority of cases of infectious encephalitis. Viral infections in the central nervous system (CNS) can alter homeostasis, induce neurological dysfunction, and result in serious, potentially life-threatening inflammatory diseases. Viruses that can cause acute encephalitis include rabies virus, herpes simplex virus (HSV), flaviviruses such as the West Nile virus (WNV) and Japanese encephalitis virus (JEV), and enterovirus including polioviruses, coxackie viruses, and echoviruses.
In healthy individuals, the number of lymphocytes that traffic into the CNS is very low and tightly controlled by the highly specialized blood-brain barrier (BBB). However, neurotrophic viruses can strategically cross the barrier systems of the CNS. The restricted expression of MHC antigens and the nonrenewable nature of the neuron cells offer additional challenges for the immune system to detect and to respond to viral infections of the CNS. The actual mechanism by which the virus reaches the CNS is not entirely defined. It may be possible that Langerhan cells or macrophages, as primary target cells, may be infected with viruses and then pass into the lymph node closest to the site of infection (2). These infected innate cells can then subsequently enter into the circulation following a brief replication in the lymph node via the efferent lymphatic system and the thoracic duct. This results in viremia, as the viruses ultimately reach the CNS (3). Recently, it has been reported that signaling through Toll-like receptors plays a key role in crossing the BBB by the encephalitis-causing viruses and influence the development ofneurological disorders (4). Although encephalitis caused by neurotrophic viruses is well-characterized in the evidence of immune system recognition and the presence of inflammatory components during the neuropathological changes (5), there are only a few studies describing the role of CD4+ helper T cell (Th) subsets, in particular the CD4+CD25+Foxp3+ Treg, during the progression of an infection caused by neurotrophic viruses (6). The purpose of this review is to provide the outline of accumulated evidences focused on their role in encephalitis caused by neurotrophic viruses.
ACUTE VIRAL ENCEPHALITIS WITH SPECIAL EMPHASIS ON JAPANESE ENCEPHALITIS
Emerging diseases are defined as diseases that have newly appeared as a threat to public health or have existed previously but are rapidly increasing in incidence or in geographical range (7). The mosquito-borne flavivirus, rising in incidence by global warming and modern transportation, is one of the most important examples of emerging and resurging diseases of global significance (8). Mosquito-borne flaviviruses with rapidly expanding ranges includes WNV, JEV, as well as the dengue virus. Notably, the introduction of WNV into North America from the Middle East (9-11) and the spread of JEV into India, Nepal, and northern Australia (12-15) are typical examples of these mechanisms. The majority of flaviviruses are zoonoses that depend on animal species other than human for their maintenance in nature, with the notable exception of the dengue virus. Humans are usually incidental and dead-end hosts that do not contribute to the natural transmission cycle. When introduced in New York, probably through air transport, WNV infected many susceptible mosquitoes and bird species to establish an endemic cycle and spread rapidly across America (9-11). Similarly, JEV is now rapidly expanded into previously unaffected regions, such as Indonesia, Pakistan, and the northern Australia (12-15). Furthermore, southern California of the United States is at particular risk for the introduction of JEV, due to its large size and function as a hub for international travel and commerce with Asia, which is a potential risk with high introduction frequency of mosquitoes infected with JEV. Similarly to WNV, if JEV is introduced into the U.S., the virus might get established rapidly, due to the large number of susceptible mosquito vectors and vertebrate hosts.
Nowadays, it is estimated that 30,000 to 50,000 cases of JE occur each year, resulting in 10,000 to 15,000 deaths. However, the actual number of cases may be underestimated due to social and political problems (16,17). Transmission occurs primarily via Culex tritaeniorrhyncus and Culex vishnui mosquitoes, which also transmit WNV. Water birds, including herons and egrets, serve as natural reservoirs and domestic pigs as an important amplifying host (17-21). JEV targets the CNS, clinically manifesting with fever, headache, vomiting, signs of meningeal irritation, and altered consciousness leading to high mortality and persistent neurological sequel in the survivors (22). In many cases, JEV is probably not directly involved in the destruction of brain tissue but may cause damage indirectly by abnormal activation of cell-mediated immune responses as well as activation of microglia and astrocytes (23). Indeed, it has been reported that various pro-inflammatory mediators like IFN-α, TNF-α, MIF, IL-8, IL-6, RANTES, COX-2, IL-1β, MCP-1 are elevated during a JEV infection (23,24). Moreover, high mortality rates were associated with increased concentrations of cytokines in sera and CSF of the victims (25). Also, the protective role of other important anti-inflammatory mediators, i.e. IL-4, IL-10, and iNOS, was demonstrated (26).
INVOLVEMENT OF TLRs IN THE PROGRESSION OF ACUTE VIRAL ENCEPHALITIS
Overview of TLR family
TLRs were first described in Drosophila on the basis of their homology to the protein Toll. A search for homologous proteins in mammals revealed the TLRs. TLRs are Type-I transmembrane pattern recognition proteins with a variable number of N-terminal leucine rich repeats (LRRs) followed by a cysteine-rich domain, a transmembrane (TM) domain, and an intracellular Toll/IL-1 receptor (TIR) domain. TLRs are crucial in the innate immune response to microbial pathogens, where they recognize and respond to pathogen-associated molecular patterns (PAMPs), which lead to the activation of intracellular signaling pathways and altered gene expression. In mammals, TLRs function as intermediates by interacting with products of infectious agents and then transmitting the signals to a cascade of adapters and kinases that ultimately lead transcription of pro-inflammatory cytokines (27). These cytokines activate innate immune cells (eg. macrophages, dendritic cells, NK cells, and neutrophils), which in turn leads to the activation of the adaptive immune system. Intracellular and extracellular TLRs can recognize a wide range of viruses leading to the production of various cytokines. Of these, the most critical in the host defense mechanism against viruses includes the type-I interferons (IFN), IFN-α and IFN-β (28).
Most mammalian species have been reported to express between ten and fifteen type of TLRs. The TLR family can be divided into five subfamilies: TLR2, TLR3, TLR4, TLR5, and TLR9. TLR2 subfamily is composed of TLR1, TLR2, TLR6, and TLR10. TLR2 forms heterodimers with TLR1, TLR6 (29-34) and probably TLR10 (30), and each TLR2-complex heterodimer has a distinct ligand specificity. TLR2 recognizes a wide range of PAMPs such as lipoproteins, lipoteichoic acid (from Gram-negative bacteria), lipoarabinomannan (from mycobacteria), glycosylphophatidylinositol anchor (from T. cruzi), phenol-soluble modulin (from S. epidermis), zymosan (from fungi), and glycolipids (from Treponema maltophilum) (29-33). TLR2 can also recognize viruses, including the measles virus (MV), human cytomegalovirus (HCMV), and hepatitis C virus (HCV) (35,36). TLR3 forms a homodimer and recognizes the viral double stranded RNA (dsRNA), which is generated during the viral replication, and induces IFN-α/β synthesis leading to various anti-viral effects and immune responses by the host (30-33). TLR4 forms a homodimer and recognizes lipopolysaccharide (LPS) from Gram-negative bacteria. This recognition process is enhanced by LPS-binding protein (LBP), which carries LPS to the CD14 molecule, where it is then presented to the MD-2-TLR4 complex (29-33). The TLR4 complex also recognizes a few other bacterial PAMPs including LTA. Moreover, the TLR4 complex recognizes viruses such as the respiratory syncytial virus (RSV), hepatitis C virus (HCV), and mouse mammary tumor virus (MMTV) (29-33,36,37). TLR5 can form a homodimer as well as a hetrodimer with TLR4. TLR5 recognizes bacterial flagellin of both Gram-positive and Gram-negative bacteria (38). TLR5 is expressed on the epithelial cells of the airways, intestine, and urogenitial tract. Interestingly, expression of TLR5 on the intestinal epithelium is polarized such that TLR5 is expressed only on the basolateral side of the cell (39). TLR7 recognizes SYNTHETIC immunomodulators such as imidazoquinolone compounds, which can be used against human papillomavirus (HPV) infections (32,33). TLR9 recognizes synthetic CpG oligonucleotides and the unmethylated CpG motifs in bacterial and viral DNA, initiating a signaling cascade leading to the production of proinflammatory cytokines (29-33,40).
Recognition of encephalitis-causing viruses by TLRs
Only limited information exists on the role of TLRs and their signaling in neurological diseases caused by viral infections. The majority of known studies involve encephalitis caused by the flavivirus infection such as WNV and JEV. A protective role for TLR7 and MyD88 in WNV infection has been reported, as mice deficient in these molecules were more vulnerable to WNV-induced encephalitis (41,42). Both TLR7- and MyD88-deficient mice showed a defect in leukocyte migration to the WNV-infected tissues, which correlated with decreased levels of IL-23. Paradoxically, systemic levels of pro-inflammatory cytokines and type-I IFN were higher in TLR7-deficient mice than wild-type mice (41), indicating that the abrogation of the TLR7 pathway had little systemic impact on type-I IFN production after WNV infection. In contrast, little effect on the systemic type-I IFN responses was observed in MyD88-deficient mice (41). The varying roles of TLR3 molecule in WNV-induced encephalitis have also been demonstrated. TLR3-deficient mice exhibited improved survival rates after WNV infection, likely due to decreased inflammatory cytokine responses that may induce decreased BBB permeability and early virus entry in the CNS (4). On the other hand, TLR3 demonstrated its protective role by restricting viral replication in neurons infected with WNV (43). Thus, the exact contribution of TLR3 in WNV infection remains controversial but likely involves both cell-intrinsic and -extrinsic pathways. In a recent study, JEV induced a functional impairment of dendritic cells (DCs), thereby dampening the CD4+ and CD8+ T cell responses that would normally be effective in fighting off the JEV infection (44). The functional modulation of DCs by JEV infection appeared to be mediated through the pan-adaptor molecule involved in TLR signaling, MyD88, both in dependent and independent manners. This indicates that TLRs may be involved in JEV pathogenesis. However, the role of each TLR molecule in JEV neuropathogenesis needs to be elucidated.
HSV is the most commonly diagnosed virus of sporadic encephalitis in humans. HSV-1 is usually acquired during childhood and often presents as a self-limiting pharyngitis. A subsequent reactivation of latently infected HSV-1 is associated with perioral lesions, termed "fever blisters." In the neonates, HSV-1 and HSV-2 cause very different diseases manifested by sepsis-like symptoms including blood pressure instability, disseminated intravascular coagulation, sometimes shock, and lethal encephalitis (45). Several TLR molecules are involved in the recognition of HSV infection. It has been reported the level of TLR2 expression increases in the brain in response to peripheral challenge with TLR2 bacterial ligands (46). HSV-1 induces inflammatory cytokines through TLR2 in both TLR2-transfected cell lines and mice (47). In vivo studies have demonstrated that TLR2-deficient mice have a blunted cytokine response and these mice are much less susceptible to normally lethal HSV1-induced encephalitis (47), which suggests that induction of inflammatory cytokines through TLR2 may lead to encephalitis (47). HSV is a double-stranded DNA (dsDNA) virus known to have a double-stranded RNA (dsRNA) intermediate usually recognized by TLR3. Upon TLR3 recognition of the dsRNA derived from HSV-1 replication, the NF-κB pathway is activated, leading to production of type-I IFNs (48), which are crucial for HSV clearance (49). A recent study showed that the synthetic TLR3 agonist has protective effects in lethal HSV-1 encephalitis (50). Also, TLR3 is expressed in the CNS, where it is required to control of HSV-1 replication (51). A genetic study reported a dominant-negative TLR3 allele in otherwise healthy children with HSV-1 encephalitis (51). These reports suggest that both the TLR3 response and the evolutionary maintenance of TLR3 might be involved in HSV-1 encephalitis (50,51). Moreover, TLR3-mediated immunity is essential for protection against HSV-1 in the CNS during the primary infection in childhood (52). It was also suggested that TLR3 has largely redundant functions for responses to dsRNA and HSV-1 in various leukocytes, probably accounting for redundancy of TLR3 responses in the host defense against HSV-1 outside the CNS (52). Since HSV DNA also contains CpG motifs that can be recognized through TLR9 (53,54), interferon-producing cells and plasmacytoid DCs are activated through the TLR9 signaling pathway (53,54).
Finally, rabies virus, another cause of neurological disease cause, is known to upregulate IFN-β, chemokines (CCL-5, CXCL-10) and inflammatory cytokines (IL-6, TNF-α, IL-1β) for TLR3-positive neurons (55). The activation of the TLR3 signal pathway indicates a possible role for TLR3 signaling in the innate recognition of rabies virus infection (55). In support of this, TLR3-deficient mice have also been reported to have reduced susceptibility to infections with the rabies virus (56).
THE ROLE OF CD4+ Th SUBSETS IN ACUTE VIRAL ENCEPHALITIS
Lineage decisions of CD4+ Th subsets
Naive CD4+ Th cells, upon encountering their cognate antigens presented by professional antigen-presenting cells (APCs), differentiate into effector cells that are characterized by their cytokine production profiles and immune-regulatory functions. The heterogeneity of effector T cells was discovered two decades ago, at which time they were divided into two subsets, named Th1 or Th2 cells (57). Th1 differentiation requires IL-12 and the transcription factors STAT4, STAT1 and T-bet (58), whereas Th2 differentiation requires IL-4 and the transcription factors STAT6 and GATA3 (59). A third effector subset in addition to Th1 and Th2 cells has been identified in recent years, the Th17 subset, the differentiation of which is induced by the combination of TGF-β and IL-6 (59,60). Recently, IL-21 was reported as an autocrine factor induced by IL-6 to regulate Th17 cell differentiation (60,61). STAT3, which is downstream of IL-6 and IL-21, is essential for RORγt and RORα expression and Th17 cell differentiation (61). Additional T cell subsets were also discovered and studied, including T follicular helper (Tfh) cells and IL-9-expressing "Th9" cells. In addition to these effector CD4+ Th subsets, there are regulatory subsets that help maintain immune homeostasis by suppressing the effector T cell responses. This helps restrain unwanted immune responses, thus preventing chronic inflammation and immune hyper-responsiveness (i.e., allergies). The thymus-derived natural regulatory T (nTreg) cells represent a unique subpopulation of CD4+ T cells and express the Foxp3 transcription factor, which is the hallmark of nTregs. Expresion of Foxp3 is essential for the Treg function in this regulatory subset (62). TGF-β has also been shown to be an important cytokine for the maintenance of peripheral nTreg cells. Additionally, naive T cells in the periphery can be induced to express Foxp3 in the presence of TGF-β, thus becoming inducible Treg (iTreg) cells that exhibit a suppressive phenotype similar to the nTreg cells (62). Lineage fate decisions of CD4+ Th subset are instructed by distinct environmental cytokines which signals through STAT or other inducible but generally ubiquitous transcription factors. Though extensive cross-regulations among lineage-determining transcription factors exist, a growing body of evidence suggests that Th cell lineage commitment can be plastic in certain circumstances.
The role of CD4+ Th subsets in viral encephalitis with special emphasis on Treg and Th17 cells
The presence of the BBB in the central nervous system that restricts the entry of cells and proteins, the restricted expression of MHC antigens, and the nonrenewable nature of the neuronal cell population offer challenges to the immune system for viral clearance and increase the chance for viral persistence. However, vigorous immune responses are mounted rapidly within the CNS against both self and exogenous antigens in the face of an antigenic challenge. The lymphocyte subsets that constitute the protective T-cell responses within the CNS have not been fully characterized. During viral infection, most CD4+ T cells isolated from the virus-targeted organs are of the Th1 type (63), and Th1 cytokines, such as IFN-γ, display strong antiviral function and antagonize the development of Th17 cells (64). In contrast to this protective strategy, a virus may be able to evade antiviral type-I and -II IFN responses (65), facilitating its persistence in the host by inducing elevated levels of IL-17-producing CD4+ and/or CD8+ T cells. This is particularly true in case of chronic viral infections, for example in the Theiler's murine encephalitis virus (TMEV) infection (66). However, Th1 cells may also play a harmful role in viral encephalitis, rather than their host-protective functions which depend on inducing an antiviral cytokine environment. In experimental autoimmune encephalitis, for instance, myelin-reactive Th1 cells in the absence of the IL-17+ Th17 cells show profound inflammatory activity (67). They can access the non-inflamed CNS, establish EAE, and facilitate the entry of Th17 cells to the CNS during EAE. Th2 cells are known to mediate humoral immunity against extracellular pathogens. Humoral immunity driven by B cells with Th2 cell leads to the inhibition of viral replication and spread in the initial stages. For example, virus neutralizing antibody-mediated inhibition of JEV replication leads to the inhibition of the cytopathic effects of the virus and hence less tissue damage in Japanese encephalitis (68). CD4+ Th2 cells may provide a protective role against viral encephalitis during infection with neurotrophic virus. JEV was known to produce CD4+ Th2 responses associated with immunoprotection against acute viral encephalitis in adoptive transfer model studies (69).
IL-17-producing Th17 cells are apparently involved in inflammatory tissue damage, leading to the pathogenesis of various autoimmune diseases (67,70). Prior to the discovery of Th17 lineage of CD4+ T cells, Th1 cells were thought to be implicated in the development of autoimmune diseases. However, more recent lines of evidence suggest that Th17 cells, rather than Th1 cells, are the true culprits in the induction and progression of many autoimmune diseases. Indeed, IL-17 expression has been detected in the serum and target tissues of patients with various autoimmune diseases such as RA, multiple sclerosis (MS), and systemic lupus erythematous (SLE). Similarly, the production of IL-17 has also been reported in several viral infections, for example, HIV infection in humans (71) and herpes simplex virus (72) and respiratory syncytial virus infections (73) in rodents. However, a persistent viral infection (eg. TMEV infection) preferentially induces the development of Th17 cells, and in turn, these cells uniquely promote the viral persistence via IL-17 by inhibiting apoptosis of infected cells as well as by desensitizing target cell killing by T effector cells (66).
Treg, a subset of CD4+ T cells, are now well-recognized for their modulatory functions and play pivotal roles in maintaining immune homeostasis and preventing autoimmune diseases (74,75). The outcome of viral infections is dependent upon complex interactions between the pathogen and the balance of pro- and anti-inflammatory immune responses by the host. Excessive T cell responses have been implicated in disease in mice infected with lymphocytic choriomeningitis virus (LCMV), herpes simplex virus, or respiratory syncytial virus, with tissue damage during the process of virus clearance (76-78). To minimize excessive pro-inflammatory responses, anti-inflammatory cells, such as regulatory T cells and the IL-10 producing Tr1 cells (79), are induced during a viral infection. An appropriate anti-inflammatory response will prevent disease caused by immune responses without adversely affecting virus clearance.
In healthy individuals, lymphocytic trafficking into the CNS is very low and tightly controlled by the highly specialized BBB. However, several pathological conditions of the CNS, such as viral or bacterial infections or inflammation-mediated disorders, induce circulating lymphocytes to cross the BBB and gain access to the inflammatory foci (80). This may be a critical step for Treg to perform their biological function in inflammatory diseases of the CNS. Alternatively, the Treg function can be mediated by their soluble factors, even without their actual extravasation to the infected sites. Recent studies revealed that several viruses belonging to the Flaviviridae family, such as the classical swine fever virus (CSFV), dengue virus (DV), JEV, and Yellow fever virus (YFV), infected DCs and altered the cell phenotype and function (81-83). Furthermore, Aleyas et al. (2009) recently reported that the Beijing-strain of JEV replicates both in bmDCs and macrophages and induced the functional impairment of DCs through MyD88-dependent and independent pathways, which subsequently led to poor CD4+ and CD8+ T cell responses (44). These findings suggest that the virus-induced alteration of DCs is likely a cause for immunosuppression mediated by Tregs. The neuronal protective effects of Tregs were found to play a role in several viral-encephalitis such as human HIV-1 encephalitis (84), Japanese encephalitis (85), and West Nile encephalitis (6). In Japanese encephalitis, the JEV P3 infection of DCs led to an expansion of CD4+ Foxp3+ regulatory T cells (Treg) with immunosuppressive potential. This suggests that the JEV-induced alteration of DCs may have initiated the immunosuppression by iTregs (85). Also, CD4+ Foxp3+ Tregs, expanded by the WNV infection, may regulate the development of virus-caused encephalitis in humans and mice (6). However, further studies will be required to elucidate how Tregs ameliorate clinical encephalitis after a neurotrophic virus infection.
PLASTICITY OF Th17 AND Treg CELLS AND THEIR BALANCE IN VIRAL ENCEPHALITIS
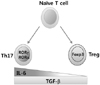
The balance between Th17 and Treg is crucial for immune homeostasis. The plasticity in the Th17 and Treg developmental programs play a pivotal role in maintaining immune homeostasis, allowing the regulation of anti-inflammatory Tregs and pro-inflammatory Th17 cell differentiation from naïve Th precursors. Although nTregs develop during thymic selection through a TGF-β-independent mechanism, the extrathymic development of iTregs is TGF-β-dependent. Development of Th17 cells is also TGF-β-dependent, but additional coordinate signaling by IL-6 produced from DC and other cells activated by microbial products, is required in concert with TGF-β to induce Th17 differentiation. In other words, priming of naïve CD4 T cells by an antigen in the presence of active TGF-β promotes the development of iTregs in the absence of pro-inflammatory signals from the innate immune system, whereas Th activation in an environment where both active TGF-β and IL-6 are available promotes Th17 development. Because TGF-β suppresses Th1 and Th2 differentiation (86), iTreg and Th17 development are favored in its presence. The transcription factor central to Th17 differentiation is an isoform of the retinoic acid-related orphan receptor γ, RORγ, which is expressed in T cells (RORγt) (87) and directs the differentiation program for pro-inflammatory IL-17+ T helper cells. A second member of the ROR family, RORα, is also associated with the Th17 development program (61), but its contribution appears dispensable. On the other hand, the transcription factor central to Treg differentiation and function is Foxp3. Interestingly, naïve T cells stimulated with TGF-β alone were found to upregulate both Foxp3 and RORγt. However, as Foxp3 is able to associate with RORγt and inhibit RORγt transcriptional activation, treatment of naïve T cells with TGF-β led exclusively to Treg differentiation (88). Therefore, the balance of TGF-β and IL-6 signaling might determine the differentiation of iTregs or Th17 cells through an antagonistic competition of Foxp3 and RORγt (Fig. 1).
Several studies recently reported that all-trans retinoic acid (at-RA), derived from Vitamin A, can potently inhibit Th17 development and promote iTreg development, at least in part by antagonizing the effects of IL-6 (89-91). This implies that interactions between Foxp3 and ROR factors could be directly or indirectly modulated by the binding of retinoic acid to the retinoic acid receptor, RAR, although specific mechanisms by which this might occur are unclear. Thus, IL-6 acts as a potent pro-inflammatory cytokine in T cells through promotion of Th17differentiation and inhibiting Treg differentiation, indicating that modulating IL-6 levels may normalize the balance between Th17 and Treg in viral encephalitis and may alleviate clinical severity.
CONCLUSION AND FUTURE PERSPECTIVES
As illustrated in this review, the studies on the immune-pathological progression of acute viral encephalitis caused by neurotropic virus have been accumulated gradually in the recent years, along with the discovery of key molecules that control the development of acute encephalitis caused by infection with the neurotrophic virus. Notably, mosquito-borne viruses that induce acute encephalitis are becoming pandemic infectious agents due to the changes in the ecosystem, global warming, and modern transportation. Paradoxically, the protective immune responses in the periphery that reduce viral burden may generate inflammatory mediators that subsequently alter the permeability of the BBB to allow free virus and/or virus-infected leukocytes to enter the CNS. The acute encephalitis caused by neurotrophic viruses is a complex immune-pathological phenomenon played by molecules related to innate and adaptive immunity. For the migration of peripheral leukocytes into the CNS, a gradient of chemokines should be established. Thus, it will be interesting to define the molecules that control the entry of leukocytes into CNS, such as TLRs and chemokines/chemokine receptors. Furthermore, the regulation of fatal neuro-inflammation caused by viral infection will be achieved by suppressive cellular mediators, such as CD4+Foxp3+ Tregs and CD11b+Gr-1+ myeloid-derived suppressive cells (MDSCs). However, despite the pathological importance of acute viral encephalitis, the immune-pathological mechanisms occurring during the progression of encephalitis and roles played by CD4+ Th subsets, especially IL-17-producing Th17 and CD4+Foxp3+ Treg cells, still remain undefined. Therefore, the future challenge will be to unravel the functions of molecules related to such cellular subsets in the proper host context. An improved understanding of the molecular signals that govern the functions of regulatory cell components may advance the development of therapeutic approaches that effectively treat acute viral encephalitis.
 XML Download
XML Download