

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Glia
"Glia" refers to a diverse set of specialized cell types that are found both in the peripheral nervous system (Schwann cells, satellite glia, perineural glia) and in the central nervous system (CNS) (astrocytes, oligodendrocytes, microglia, and perivascular glia) (1). Glial cells constitute 70% of the total cell population in the brain and spinal cord. Glial cells can be subdivided into two primary categories: microglia, comprising 5% to 10% of the glial population, and macroglia, which include astrocytes and oligodendrocytes (2). Glial cells that were, up to several years ago, considered the "forgotten brain cells," or neglected stepchildren of neuroscience, now act as orchestrators in the tetrapartite synapse, and control of their activation state is crucial to the integrity of CNS function. Recent technical advances and increased interest in elucidating the role of non-neuronal cells of the CNS in both the physiological and pathophysiologial processes have catapulted these cells into the forefront of neuroscience research. Glial cells do not conduct nerve impulses, and they provide structural support for the brain, assisting in nervous system development, repair and maintenance, supplying nutrients and biosynthetic products to neurons, imparting metabolic functions to neurons, destroying and removing injured and dead neurons and, finally, regulating the neuronal microenvironment. The well known quote 'the neuron is the structural and functional unit of the brain' has been challenged by a wealth of findings which have, to the contrary, introduced the 'neuronal-glial complex' as the structural and functional unit of the brain. Glia regulate brain vasculature and the blood-brain barrier, modulating ischemia and migraines. Moreover, they are important in the repair of neurons after injury and also contribute to neuropathology in neurodegenerative diseases. Microglia can either protect or damage neurons depending on where and how they are activated. Microglia are chronically engaged in repairing minor insults and that clinical diseases are observed only when these repair efforts fail. Fully activated microglia are detrimental to neurons, but other stages in the sequence of reactive states may improve neuronal survival by releasing neurotrophic factors or by removing excess glutamate from the extracellular space (3). Glial cells, which include oligodendrocytes, astrocytes, and microglia, have been found to play key roles in neuroinflammation and neuropathic pain. Given that less is known about the involvement of oligodendrocytes, this paper will focus primarily on the role of astrocytes and microglial cells in neuroinflammation and neuropathic pain. In view of the neuroinflammation and neuropathic pain processes, activation of glial cells in the spinal dorsal horn, especially microglia and astroglia, plays a predominant role. Furthermore, astrocytes and microglia are known to play a role in the development, spread, and potentiation of neuropathic pain (4-12).
Neuroinflammation
Neuroinflammation is a normal and necessary process. In the acute phase after injury, neuroinflammation is tightly controlled. In its chronic phase or when directed against normal tissue (in an autoimmune response), neuroinflammation is detrimental when it manifests itself as multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), various types of dementia, Huntington's disease, or other diseases (13-16). A wide range of neurodegenerative diseases, including those affecting the CNS, such as Alzheimer's disease (AD), Parkinson's disease (PD), ALS, and MS, are associated with chronic inflammation (17-21). Although inflammation may not be the initiating factor, emerging evidence in animal models suggests that sustained inflammatory responses involving microglia and astrocytes contribute to disease progression (22). The activation of perivascular microglia and endothelial cells (lining the capillary bed), astrocytes (making up the blood-brain barrier), parenchymal microglia and astrocytes by the means of stresses, leads to the subsequent production of cytokines, cellular adhesion molecules, chemokines, and the expression of surface antigens that enhance a CNS immune cascade. If left unchecked, this neuroimmune activation can lead to the trafficking of leukocytes into the perceived area of injury as a mechanism for neuroprotection. Therefore, neuroinflammation can be defined as the infiltration of immune cells into the site of injury in response to damage to the peripheral or CNS (23).
Neuropathic pain
Neuropathic pain, initiated or caused by primary lesions or dysfunction in the CNS (brain and spinal cord) or the peripheral nervous system (nerves outside the brain and spinal cord), having occurred following viral infection, trauma, certain medications, or metabolic insults, and many diseases such as MS and stroke, is especially problematic because of its severity, chronicity, and resistance to simple analgesics (24). The possible mechanisms of neuropathic pain could be classified as: a) chemical excitation of non-nociceptors, b) recruitment of nerves outside the site of injury, c) excitotoxicity, d) excess sodium channels, e) ectopic discharge, f) central sensitization maintained by peripheral input, and g) sympathetic involvement.
THE ROLE OF GLIA IN NEUROINFLAMMATION
The role of microglia in initiating or promoting inflammatory processes in the CNS by facilitating the recruitment of peripheral immune cells has been well documented (25-27). During the neuroinflammatory process, microglial cells release proinflammatory mediators such as cytokines, matrix metalloproteinases (MMP), reactive oxygen species (ROS), and nitric oxide (NO). For a long time, glial cells have been considered to merely support the neuronal environment. However, the innovative research in the field of neuroscience has strongly propelled glial cells as new players in neuroinflammation and neuropathic pain. Neuroinflammation is a characteristic feature of both acute and chronic CNS disorders and is a process that results primarily from the presence of chronically activated glial cells (astrocytes and microglia) in the brain, and is a common feature of several neurodegenerative conditions. Activated glia release a variety of neuroexcitatory substances that potentiate neurotransmission, especially proinflammatory cytokines. Blocking glial activity may be a novel way of controlling neuropathic pain. Neuroinflammation induces a complex and dynamic change in glial cell phenotypes. One of the first cell types to respond are microglial cells, which retract their processes and migrate towards the site of injury, where they release proinflammatory cytokines such as IL-1β, TNF-α, and IL-6 (28-30).
THE ROLE OF GLIA IN NEUROPATHIC PAIN
Neuropathic pain is a debilitating condition that affects millions of individuals worldwide. It is now thought that solely considering neuronal activity provides an incomplete understanding of the creation and maintenance of chronic neuropathic pain (31). Glia have recently emerged as key contributors to pathological and chronic pain mechanisms and are emerging as a new target for drug development (32,33). Spinal cord glial activation seems to be a common underlying mechanism that leads to pathological pain in a number of pain syndromes with dramatically different aetiologies (for example, diabetic neuropathy, chemotherapy-induced neuropathy, peripheral nerve inflammation and trauma, and spinal cord inflammation) (34). Upon activation, both the astrocytes and microglia respond to and release a number of signaling molecules which have protective and/or pathological functions. These include, among others, the classic immune signals: cytokines and chemokines. The role of glia in the CNS is intimately integrated with the functions of the other players in the tetrapartite synapse composed of pre- and post- synaptic neurons, astrocytes, and microglial cells. In trauma or disease states, the spinal glia become activated and the dorsal horn neurons become hyperexcitable, contributing to sensitized neuronal-glial circuits. The maladaptive spinal circuits directly affect synaptic excitability, including the activation of intracellular downstream cascades, that results in enhanced evoked and spontaneous activity in dorsal horn neurons leading to the development of abnormal pain syndromes (35).
INTERLINKING INFLAMMATION AND NEUROPATHIC PAIN
Inflammation is a component of the body's wisdom. Inflammation is the body's foremost response to an injury or disease. Webster defines inflammation as "redness, swelling and fever in a local area of the body, often with pain and disturbed function, in reaction to an infection or to a physical or chemical injury." Symptoms of inflammation can range from mild aches to sharp, wrenching pain that may take one's breath away. Inflammation in any part of our body is coupled with pain, due to the release of inflammatory mediators such as prostaglandin E2 (PGE2), the proinflammatory cytokines TNF-α and IL-1β, and nerve growth factor (NGF). These mediators, produced by non-neural cells or immune cells, can stimulate nociceptor terminals in the peripheral tissue to increase pain sensitivity (36,37). Inflammation also occurs in the CNS after brain trauma, brain infection, and in neurodegenerative diseases, such as AD, PD, and MS. This so-called neuroinflammation is characterized by the activation of glial cells (especially microglia and astrocytes) in the CNS and is an important contributor to the development of neurodegeneration by releasing inflammatory mediators from glial cells (38,39). This glia-mediated neuroinflammation also plays an important role in pain control under pathological conditions.
Although multiple conditions may generate neuropathic pain, a common underlying mechanism is the presence of inflammation at the site of the damaged or affected nerve(s). This inflammatory response initiates a cascade of events resulting in increased local perfusion, increased capillary permeability, and the concentration and activation of innate immune cells at the site of tissue injury, irritation, or infection. Immunoactive substances, such as cytokines, neurotrophic factors, and chemokines, released at the site of injury have local actions and can initiate a systemic immune response. The resultant neuroinflammatory environment can cause the activation of microglia and astrocytes located in the spinal cord and brain, which appear to play a prominent role in nociception (40).
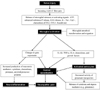
Current research suggests that microglia are involved in the early development of neuropathic pain, whereas astrocytes function to sustain neuropathic pain (12,41-43). The activation of spinal cord glia is both necessary and sometimes even sufficient for the development of persistent pain states associated with various etiologies, including diabetic neuropathy, chemotherapy-induced neuropathy, peripheral nerve inflammation and trauma, and spinal cord inflammation (44-46). Both spinal astrocytes and microglia activate mitogen-activated protein kinases (MAPKs) to induce the synthesis and release of proinflammatory cytokines, such as IL-1β, IL-6, TNF-α, PGE2, and NO (47,48). There is ample evidence that both astrocyte and microglia activation lead to pro-inflammatory responses with pathological effects, such as neuronal hyperexcitability, neurotoxicity and chronic inflammation. Fig. 1 illustrates the consequential neuroinflammation and neuropathic pain subsequent to the neuronal injury via the glial cell (microglial and astrocytic) activation.
GLIA IN FORMALIN TEST & COMPLETE FREUND'S ADJUVANT (CFA)-INDUCED INFLAMMATORY PAIN MODEL
Animal models of inflammatory pain have been widely used to study the mechanisms of tissue injury that induced persistent pain. The formalin test is the most predictive of the models for acute pain, it is predominantly used on rats and mice and involves moderate, continuous pain generated by injured tissue. In this way it differs from most traditional tests of nociception which rely on brief stimuli of threshold intensity. Formalin injected beneath the footpad of a rat, mouse, or cat produces two phases of nocifensive behavior, characterized by the licking and flinching of the paw, that are separated by a short period of quiescence in which there is no apparent pain behavior (49,50). The first or "acute" phase typically occurs in the first 5 min; the second starts from 15 min and lasts about 40~60 min after injection. The early phase seems to be caused predominantly by the direct activation of both the low-threshold mechanoreceptive and the nociceptive primary afferent fibers due to the peripheral stimulus (51). The late phase, also called the tonic phase, appears to be dependent on the combination of an inflammatory reaction in the peripheral tissue and functional changes in the dorsal horn of the spinal cord, popularly called central processing. These functional changes seem to be initiated by the C-fiber barrage during the early phase (52).
Complete Freund's adjuvant (CFA)-induced inflammatory pain is very commonly used as a model for chronic inflammatory pain. CFA contains heat-killed or inactivated and dried mycobacteria, which is a primary agent responsible for stimulating antibody production, but has also been attributed to a number of undesirable side effects (53). This assay has a good track record for predicting the effectiveness of compounds as analgesic or anti-hyperalgesic agents as well as defining the mechanism behind inflammatory pain. CFA is injected into one hind-paw of the animal. The CFA injection in the footpad produces localized inflammation and persistent pain (54,55). Thermal hyperalgesia or mechanical allodynia associated with the inflammation are assessed by determining the hind-paw withdrawal latency. After the CFA injection into the footpad, cutaneous inflammation appears in minutes to hours and peaks within 5~8 hours. The average time of onset is 2~6 hours and persists for approximately 1~2 weeks. The edema peaks around 24 hours after injection (56). For the induction of hind paw inflammation, mice receive (ipl.) injection of 10µl of CFA (diluted with PBS, 2 mg/ml Mycobacterium tuberculosis). The CFA produces dose-dependent inflammatory responses. Behavior testing in CFA-injected mice is done by mechanical sensitivity testing using von Frey hairs and heat sensitivity testing.
Peripheral formalin injection induces two stages of microglial activation: p38 activation in spinal microglia plays key roles in central pain modulation in formalin test for both the early acute phase and the late secondary long-term pain state (57). These unique properties of spinal microglial activation in an animal model of pain will potentially help to further understand the contributions of spinal microglia to acute and chronic pain states. Both qualitative and quantitative analyses for the comparison of the effects of the peripheral CFA and formalin injection on spinal microglia activation showed signs of microglia activation on the ipsilateral side of the lumbar dorsal horn on days 3, 7, and 14 after the formalin injection was introduced. However, significant microglia morphological alteration was not found in the CFA model. At the injection site in the paw, the CFA injection induced considerably more inflammation than the formalin injection. Although spinal microglia might have been activated morphologically in inflammatory pain models, spinal microglia activation was not closely correlated with peripheral inflammation (58). Glial activation is a common feature of many diseases of the CNS (59). In the spinal cord, astrocytes are activated following peripheral inflammation or the nerve injury and may manifest as increased expression of astrocytic markers such as glial fibrillary acidic protein (GFAP) (60,61). The peripheral injection of CFA increased the mRNA and protein expression of the astrocytic marker GFAP in the bilateral anterior cingulate cortex (ACC). The inhibition of astroglial function by an astroglial toxin blocked the place-avoidance behavior, but not the paw withdrawal threshold, suggesting the involvement of astrocytes in the ACC as the affective component of pain. Modulating the function of astrocytes in the ACC may provide a new strategy for the prevention of chronic pain-induced emotional disturbances.
CONCLUSION AND FUTURE PERSPECTIVES
Neurons are not the only cell type in the nervous system; ~90% of the cells in our brain are glia. Glia, which until recently were thought to be passive support cells for the neurons, are now considered to be, not only the link between neuroinflammation and neuropathic pain, but also an important link between the immune and nervous systems under inflammatory and traumatic conditions. Microglia, not 'born' in the nervous system but formed by the transformation of certain white blood cells called macrophages or their precursors, monocytes, are now thought to be part of the immune system, defending the brain against infection and injury. Microglia are the macrophages of the brain and are the first responders to CNS injury, but exactly which signal triggers microglial reactivity is not fully understood. The activating signals may include changes in neuronal transmission or the appearance of NO or proinflammatory cytokines. Substantiation indicates that central glial activation depends on nerve inputs from the site of injury and the release of chemical mediators. Glia, being the secret player in the neuroinflammation and neuropathic pain, could be exploited to provide new research avenues for therapeutic pain control. Taking into account both the protective and the pathological effects of activated glia will be a key to the development of effective therapeutics. Although knowledge of the development and differentiation of glial cells has significantly increased in recent years, there are still many unanswered questions regarding the consequential neuroinflammation and neuropathic pain subsequent to the neuronal injury via glial cell (microglial and astrocytic) activation.
Therapies directed at activated glia hold promise for new approaches to intractable pain. To expedite the goal of developing new diagnostic tools and new therapies for intractable pain, it is important to allow the cross-fertilization of ideas to occur between preclinical and clinical researchers. It is high time that neuroscience research focused on the characterization of the glial phenotypes in the circumstances of inflammation, nerve injury and their correlation with pain behavior. It can lead to the development of new therapeutic strategies by targeting both spinal glial cells and bone marrow-derived macrophages for effective pain relief. Finally, it can be concluded that glial cells act as orchestrators in the tetrapartite synapse and control of their activation state is crucial to the integrity of CNS function.
 XML Download
XML Download