

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
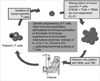
ACT is based on the transfer of ex-vivo expanded anti-tumor or anti-viral CD8 T cells into affected patients and has shown positive results in clinical trials (Fig. 1) (1). Historically, the concept of ACT was first introduced when a paper in 1964 demonstrated that the transfer of immune lymphocytes could inhibit growth of a carcinogen-induced rat sarcoma (2). The effectiveness of ACT was later enhanced by use of recombinant IL-2; it was the first breakthrough in ACT due to IL-2's ability to induce T cell stimulation and proliferation. Although recombinant IL-2 treatment need to be used carefully in clinic by toxicity such as 'capillary permeability leak syndrome' that results in major fluid retention, it had been previously approved by the Food and Drug Administration (FDA) for the treatment of metastatic renal cancer in 1992 and metastatic melanoma patients in 1998 (1).
IL-2 characteristically enhances immunological functions. In the context of cancer, IL-2 can be used as a T cell growth factor in in vitro culture, and the intravenous injection of tumor-sensitized lymphocytes grown in long term culture in the presence of IL-2 is capable of curing mice with established local and disseminated syngeneic tumor (3). Moreover, systemic administration of IL-2 with tumor-sensitized lymphocytes exhibited enhanced therapeutic potential in an established tumor model of mice (4). Later, lymphocytes grown in culture with IL-2 were referred to as lymphokine-activated killer (LAK) cells. However, administration of a high dose of IL-2 and LAK cells was not shown to be superior in survival to that of IL-2 alone when patients had metastatic cancer (5).
In 1986, Rosenberg et al. showed that an adoptive transfer of tumor-infiltrating lymphocytes (TIL) was 50 to 100 times more effective than that of LAK cells (6). Moreover, IL-2 administration in combination with TIL could overcome limitations attributed to the use of TIL only, such as lowered activation or suppression of TIL. For this reason, in most current protocols, a high-dose of IL-2 is administered intravenously. Subsequently, the first clinical trial of TIL infusion with IL-2 administration for metastatic melanoma patients was published in 1988 (7). Despite promising initial results, treatment with TILs and IL-2 resulted in objective responses in only about one third of patients with metastatic melanoma (8). Currently, the expanded TIL cultures were selected by tumor-antigens for highly active tumor-specific T cells to enhance the efficacy of ACT with TIL (9,10).
The induction of immuno-depleting condition in patients before ACT by using non-myeloablative chemotherapy (NMC) and total body irradiation (TBI) was the second breakthrough in the field of ACT (1). Previous studies have already demonstrated a correlation between immune-depleting conditions and the efficacy of ACT (11-15). For example, immuno-depleting conditions by NMC using cyclophosphamide and fludarabine provided an optimal environment for infused TILs by eliminating suppressive regulatory T cells (Treg) and reducing competition with endogenous lymphocytes to receive the signal of homeostatic cytokines such as IL-7 and IL-15. Additionally, TBI can further augment lympho-depleting condition. Using ACT in the setting of NMC was demonstrated to mediate the regression of established tumors in ~50% of malignant melanoma patients (16).
The third breakthrough in ACT occurred when the differentiation properties of T cells were defined. Effector T cells differentiating in the later stages of long-term culture were found to be less effective in in vivo tumor treatment (17). This finding challenged the methodology of using long-term culture of T cells. However, Berger et al. demonstrated that CD8 T cells derived from central memory T cells (TCM), but not effector memory T cells (TEM), persisted long-term in vivo in macaques and reacquired phenotypic and functional properties of memory T cells and occupied memory T cell niches (18). Thus, TCMs were more effective than TEMs in enhancing ACT efficacy. More recently, Gattinoni et al. also identified stem cell-like memory T cells (TSCM) in humans, which consistently express a surface marker typically found on naïve T cells. However, unlike naïve T cells, TSCMs express Sca-1, Bcl-2, IL2Rβ and CXCR3 (19). TSCMs were shown to display greater antitumor effect than TCM when adoptively transferred into tumor bearing mice, which indicates TSCMs may play a greater role in future ACTs (19,20).
These three breakthroughs appeared to significantly improve ACT efficacy (Fig. 2). However, the ACT approach is not still satisfactory and needs to be further developed to effectively treat hard-to-cure cancer patients in clinics. The next sections will enumerate novel strategies that may make the ACT approach successful for clinical purposes in the near future.
GENETIC MODIFICATION OF T CELLS
Recently, gene modification of T cells has also been utilized for enhancing the efficacy of ACT (Fig. 2). The transduction of α and β TCR was first introduced to confer reactivity against tumor-associated antigen (TAA). Genes encoding TCRs can be isolated from T cells with a high avidity for recognizing TAA. And, the retroviral or lentiviral vectors can be used to deliver TCR genes and redirect lymphocyte specificity to TAA (Fig. 3). This procedure allows the rapid production of TAA-specific T cells and has been applied to several antigens such as minor histocompatibility antigen (21), CEA (22), gp100 (23,24), MART-1 (25), p53 (26), NY-ESO-1 (27,28), WT-1 (29) and AURKA (30). The aforementioned approach was reported to be put into clinical trial in 2006 for the first time (25). In this trial, Morgan et al. reported that this method was indeed feasible by showing durable engraftment at levels exceeding 10% of peripheral blood lymphocytes of adoptively transferred genetically modified MART-1-specific T cells into 15 patients. In two of the patients, the engineered cells were maintained in the blood even 1 year after infusion and continued to demonstrate objective regression of metastatic melanoma lesions.
Despite a positive outlook of α and β TCR transduction, Johnson et al. reported the possibility of toxicity by adoptive transfer of engineered TAA-specific T cells (31). When engineered TAA-specific T cells were administered to 36 patients with metastatic melanoma, engineered cells persisted at high levels in the blood of all patients at 1 month after infusion as expected and approximately 20 to 30% of patients exhibited objective cancer regression. However, though these initial results were promising, patients also exhibited a destruction of normal melanocytes in the skin, eye, and ear and sometimes required local steroid administration to treat uveitis and hearing loss. In addition, Parkhurst et al. recently reported in 2011 that engineered T cells mediate regression of metastatic colorectal cancer but induce severe transient colitis in patients (32). Thus, highly reactive TAA-specific T cells can mediate cancer-regression, but also target rare cognate-antigen-containing normal tissue throughout the body.
Although there is no clinical report, the cross pairing of transduced TCR chains and endogenous TCR chains, which form hybrid TCRs, may mediate unexpected autoimmune reactivity (33). Thus, several reports have already suggested strategies to prevent cross pairing of transduced TCR chains and endogenous TCR chains. Firstly, hybrid TCRs, which consists of a human variable region and murine constant region, can prevent cross pairing and are overexpressed on the surface when transduced into human lymphocytes (34,35). Preferential pairing of murine constant regions and improved CD3 stability seemed to prevent cross pairing. Secondly, the introduction of an additional disulfide bond into the α-and β-chains of TCRs improves preferential pairing with each other, increases total surface expression of transduced TCR chains, and reduces cross pairing with endogenous TCR chains (29,36,37). Thirdly, mutations in the transduced TCR structure promotes selective assembly of transduced TCRs while preserving its specificity and avidity for Ag ligands (38). Fourthly, the incorporation of CD3ζ into transduced TCRs results in highly preferred pairing between TCRα:CD3ζ and TCRs:CD3ζ and also prevents TCR cross pairing with TCR-chains: CD3ζand endogenous TCR chains (39,40). Fifthly, using a vector system encoding siRNAs for endogenous TCR genes can prevent the expression of endogenous TCRs (41). In this case, it is possible to prevent the reactivity of engineered T cells to normal cells by endogenous self-recognized TCR. Finally, transduction of engineered TCR genes into γδ T cells can prevent cross-pairing with transduced TCRs and endogenous TCRs in addition to maintaining their functionality, which includes specific proliferation capacity, Ag specific reactivity, in vivo persistence, and recall response (42).
However, the use of artificial αβ TCRs has a fundamental limitation because some tumors display lower levels of MHC class I molecules, resulting in poor efficacy of ACT by engineered T cells. Thus, the chimeric antigen receptor (CAR) was introduced to compensate for this limitation. CAR is generated by joining the light and heavy chain variable regions of a monoclonal antibody, referred to as a single chain Fc (scFv) molecule, with the hinge domain, transmembrane, and cytoplasmic signaling domains derived from the CD3ζ chain or Fc receptorγ chains. CAR directly recognizes cell-surface antigens and confers specificity of engineered T cells independently of antigen processing or MHC-restricted presentation. An example of a CAR target antigen was well summarized in a review by Sadelain (43). The first generation of CARs, whose signaling domain contained CD3ζ or FcRγ, has been shown to deliver a potent T cell activation signal. However, it was not sufficient enough in subsequent activation steps in the absence of a concomitant costimulatory signal (44). As a result, the second generation of CARs contained the signal transduction domain of CD28, 4-1BB, or other costimulatory signaling domains, which enhanced the therapeutic potential of engineered T cells (44-47). The most recent third generation of CARs contains triple-fusion signaling domains to enhance the therapeutic potential of engineered T cells (48).
An additional strategy to enhance the efficacy of ACT is transduction of anti-apoptotic molecules (Fig. 3). An intrinsic death pathway can be blocked by overexpression of Bcl-2. As evidence, tumor-specific T cells overexpressing Bcl-2 by gene transduction maintained their potential to recognize their target and were resistant to apoptosis (49). Moreover, anti-apoptotic molecule, Bcl-xL, is not expressed in resting T cells, but CD28 co-stimulation can induce transient expression of Bcl-xL, leading to resistance of Fas-induced apoptosis. Therefore, Bcl-xL transduction into T cells will also result in enhanced persistence of engineered T cells in vivo with thereby potentially improving the efficacy of ACT (50). Transduction of siRNA for FAS also makes engineered T cells resist FAS-induced apoptosis (Fig. 3) (51). Secondly, CD8 T cells did not become senescent when they were transduced with human telomerase (hTERT) (52), which is responsible for ensuring that the length of telomeres is relatively consistent, and led to persistence of engineered T cells in vivo (53). Thirdly, when dominant negative TGF-βRII was transduced into T cells, the engineered T cells resisted the inhibitory effects of tumor-derived TGF-β (Fig. 3) (54). TGF-β is an immunosuppressive cytokine produced in most human tumors and is known to markedly inhibit tumor-specific T cell responses, which is one of several immune response evasion strategies of tumors. Lastly, T cells transduced with some selected costimulatory molecules, CD80 and 4-1BBL, showed the potent tumor elimination of large and systemic tumors in immunodeficient mice (55). The engineered T cells can provide agonistic costimulatory signals to tumor-infiltrating T cells (trans-costimulation) and also provoke auto-costimulation to overcome adverse tumor microenvironment.
SAFETY ISSUES OF GENETICALLY-ENGINEERED T CELLS
In 2010, one case report suggested that autologous HER2-specific CAR-engineered T cells were also detrimentally toxic (56). In the case report, following completion of NMC, a patient received 1010 HER2-specific CAR-engineered T cells intravenously. Within 15 minutes after the cell infusion the patient experienced respiratory distress and displayed dramatic pulmonary infiltrate in chest X-rays. Despite intensive medical intervention the patient died 5 days later. Serum samples of the patient after infusion showed marked increases in IFN-γ, GM-CSF, TNF-α, IL-6 and IL-10 implying that over-reactivity of HER2-specific CAR-engineered T cells on a small amount of HER2 expressed in normal tissue. This case report demonstrated that safety should be of great concern in preventing graft-versus-host disease (GVHD) when using engineered T cells.
As a consequence, transduction of suicide genes, such as the herpes simplex virus thymidine kinase (HSV-tk), is suggested to control potential GVHD cases as a safety precaution (Fig. 3) (57). Using the HSV-tk suicide gene in engineered T cells, for instance, allows for administration of GCV when GVHD occurs; GCV then induces the apoptosis of the HSV-tk transduced T cells. However, one disadvantage of HSV-tk is that it is foreign by nature, and immune responses to HSV-tk transduced T cells could lead to their premature elimination after infusion (58,59). This is supported by evidence showing that, after infusion of HSV-tk transduced engineered T cells, HSV-tk-specific T cells were detected in the blood; repeated infusion of HSV-tk transduced engineered T cells induced memory HSV-tk-specific T cells (60). Thus, when HSV-tk transduced engineered T cells persist in vivo, they are a small fraction and contain HSV-tk transgene deletion by an anti-HSV-tk immune response after infusion (61).
Other than HSV-tk transduction, there are other, alternative strategies to prevent GVHD; firstly, human thymidylate kinase (62), human caspase 9 (63), or human FAS gene (64) can be transduced into autologous T cells to induce apoptosis of transduced T cells when GVHD occurs. Secondly, Griffioen et al. has shown that transduction of CD20 into T cells as a suicide mechanism efficiently induced elimination of engineered T cells by complement dependent cytotoxicity after treatment of therapeutic anti-CD20 antibody named rituximab (65). Thirdly, to circumvent these removal strategies, the use of RNA transfer technology is suggested for the benefit of safety concern. This is supported with the finding that when RNA is transferred into primary T cells, the expression of targeted protein is transient and gradually decreasing. Using RNA encoding CIR(chimeric immune receptor) against Her-2/neu antigens, antitumor effect was showed compared with Herceptin in vivo (66).
Additional strategy for enhancing the efficacy of ACT
Several strategies were introduced to enhance the efficacy of ACT; the first method is inhibition of the immunosuppressive environment in vivo. Selective depletion of regulatory T cells and blockade antibodies of cell surface inhibitory molecules (CTLA-4/PD-1) or inhibitory cytokine TGF-β secreted from tumor cells can enhance the efficacy of infused T cells (67). The second method is to enhance the functionality of infused T cells indirectly. Additional vaccination with peptide/virus/APC containing antigens after infusion of T cells can stimulate the infused T cells (68-70). Moreover, administration of TLR agonists further enhance the efficacy of infused T cells, followed by a long-term cure of large tumors (71). In addition, alternative cytokines, such as IL-7, IL-15 and IL-21 have been tested to increase the efficacy of infused T cells instead of IL-2 (72).
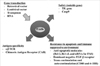
Recently, α-Galactosylceramide (αGalCer) was introduced to enhance the efficacy of ACT. When activated T cells were pulsed with αGalCer before infusion, the infused activated CD8 T cells markedly proliferated, secreted more cytokines, exhibited cytotoxic lymphocyte activity, and persisted for longer durations (73). Because αGalCer is known as a NKT cell ligand presented on CD1d (MHC class I like molecule), it induces NKT cells to secrete both Th1 and Th2 cytokines. These secreted cytokines activate a variety of other cell types, including NK cells, DCs, B cells and T cells (74). Fig. 4 suggests a possible action mechanism for αGalCer-pulsed CD8 T cells in ACT. First, αGalCer incorporated into the membrane of infused CD8 T cells is transferred into the membrane of DCs in a cell-to-cell contact manner. Subsequently, the transferred αGalCer is internalized via endocytosis into the endosomes of DCs. Thereafter, αGalCer associates with CD1d of DCs, perhaps through assistance by lipid transfer protein (LTP) (75,76). Afterwards, αGalCer-CD1d complexes are transported from the endosome to the cell surface for presentation to iNKT cells (77) where activated iNKT cells by αGalCer on DCs secrete IL-2, which enhance the proliferation of infused CD8 T cells.
CONCLUSION
Currently, ACT is one of the most effective cell therapies with objective responses in more than 50% of the cancer patients when used in parallel with IL-2 and immunodepleting regimens. Moreover, the introduction of gene modification of T cells further enhanced ACT efficacy though there remain safety concerns using this methodology. Recently, using stimulatory glycolipid pulsing on infused T cells and combination with host immune modulators have been demonstrated to significantly increase the efficacy of ACT. Therefore, the combinatory effects of several therapeutic methods including genetic modification, αGalCer-pulsing, and other immunotherapies will be used as a practical approach to treat effectively hard-to-cure cancer patient in the near future.


 XML Download
XML Download