

 ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Dengue virus (DenV) has four serotypes (DenV1, DenV2, DenV3, and DenV4), and belongs to the Flavivirus genus of the Flaviviridae family transmitted to humans by the mosquito Aedes aegypti (1-3). DenV poses a significant public health threat to 2.5 billion people at the risk of infection (1-3). Around 100 million cases of DenV infections occur annually, producing symptoms ranging from mild fever to severe hemorrhagic, potentially fatal fever (1-3). Dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS), which reportedly affect about 500,000 people per year, are potentially fatal diseases (1-3). These diseases are spreading from tropical to subtropical areas of the world by global warming, increasing travel activity, and uncontrolled urbanization (1-3). Despite global morbidity and mortality, the pathogenesis of diseases caused by DenV infection is poorly understood. Even though several factors such as viral virulence, age and genetic predisposition of the patient are implicated, the most important factor is considered to be sequential infection by different serotypes in an endemic area (4,5). While approved vaccines remain unavailable, several approaches to develop a dengue vaccine have been evaluated. These include the traditional live attenuated vaccines (6-9), recombinant subunit vaccines produced using several different host systems (10-13), chimeric virus such as yellow fever vaccine vector YF17D-based ChimeriVax (14,15) and RepliVax (16), and DNA vaccine (17,18).
All four dengue serotypes have co-circulated in most endemic countries at various times, thereby causing concurrent and/or sequential infection by multiple serotypes (19). Furthermore, there is the potential for antibody-dependent enhancement (ADE) associated with non-neutralizing cross-reactive antibodies arising from immunization with monovalent dengue virus vaccines (19). Therefore, a dengue virus vaccine should elicit protective immunity simultaneously to all four serotypes. The current approach to making a tetravalent dengue vaccine is to create monovalent vaccine candidates, and then mix these to obtain a tetravalent formulation (20-22). The application of this approach to live, replicating virus vaccine has revealed the potential for viral interference in some instances (22,23). Considering viral interference in vaccination with live viral vector vaccine, multiple primeboost vaccinations with alternating vaccine vehicles using DNA vaccine expressing the same antigen may become an effective strategy for eliciting robust immune responses to the target antigen (24). Notably, the prime-boost protocol, in which antigen-encoding DNA vaccine is administered first, followed by a boost with live viral vector expressing the same antigen, has elicited effective protective immunity in both mouse and primate models of several infectious diseases (25,26). However, some experiments claimed that priming with live viral vector vaccine and boosting with DNA vaccine induces superior immune responses against encoded antigens (27,28), which suggest that optimal prime-boost protocol to induce effective immunity may be dependent on several factors such as encoded antigens, animal species, and properties of vaccine vectors.
Various viral vectors expressing foreign antigen, such as vaccinia virus, adenovirus or Fowlpox have been used for prime-boost vaccination. DNA- and vaccinia-based vaccines for a pre-erythrocytic malaria antigen that were delivered in a prime-boost protocol induced 5- to 10-fold greater T-cell responses than each vaccine alone (29). In addition, gene-based vectors, such as replication-incompetent adenovirus, have proven particularly effective in eliciting enhanced cellular and humoral immunities compared to either agent alone (30,31). Replication-incompetent adenovirus has the ability to efficiently deliver antigens and express them at high levels in a variety of cells, making them ideal vaccine carriers (32,33). In contrast, vaccinia virus-vectored recombinants show unrestricted replication in immune-compromised individuals (34). Here, we explored the immune responses induced by alternating prime-boost vaccination using these different viral vector vaccines expressing E protein of DenV2, which is a major immunogen involved in conferring protective immunity against dengue infection. We found that vaccinia virus expressing E protein produced better responses compared to adenovirus at equivalent antigen doses, and these viral vectors elicited different patterns of CD4+ and CD8+ T cell responses against E protein when used for alternating prime-boost vaccination. Therefore, humoral and cellular immune responses induced by alternating prime-boost vaccination using live viral vector vaccine, vaccinia and adenovirus, and DNA vaccine are discussed.
MATERIALS AND METHODS
Animals and ethics statement
Female BALB/c (H-2d) mice, 5 to 6 weeks of age, were purchased from Samtako (Osan, Korea). The mice were maintained in the animal facility at Chonbuk National University under standard conditions. All experimental procedures and animal management procedures were undertaken in accordance with the requirement of the Animal Care and Ethics Committees of Chonbuk National University. The animal facility at Chonbuk National University is fully accredited by the National Association of Laboratory Animal Care.
Plasmid DNA preparation
Plasmid DNA encoding E protein of dengue virus type 2 (DenV2) under the control of the cytomegalovirus (CMV) promoter (pDE) was constructed by inserting cDNA of the E protein gene into pCI-neo vector (Promega, Madison, WI). For immunization, plasmid DNA was purified by polyethylene glycol (PEG) precipitation as described previously (35). Briefly, cellular proteins were precipitated with one volume of 7.5 M ammonium acetate followed by isopropanol precipitation of the supernatant. After PEG precipitation, the plasmid was extracted three times with phenol-chloroform and precipitated with pure ethanol. The DNA quality was checked by electrophoresis on a 1% agarose gel. The plasmid DNA concentration was measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientitific, Pittsburgh, PA). The amount of endotoxin was determined by the Limulus amebocyte lysate (LAL) test (<0.05 EU/µg). The in vivo effect of endotoxin and CpG was addressed by parallel administration of the control vector, pCI-neo.
Construction of recombinant adenovirus expressing E protein
The E1- and E3-deleted expression vector into which E protein of DenV2 is cloned was used to construct replication-incompetent adenovirus expressing E protein of DenV2 (36). E protein of DenV2 was expressed in the replication-incompetent adenovirus by cloning E protein gene under the control of the human CMV promoter. We initially constructed recombinant entry vector pENTR11 (Invitrogen, Carlsbad, CA) containing E protein gene by RT-PCR amplification and subcloning. Using LR Clonase (Invitrogen) for catalysis, the recombinant entry vector pENTR11 containing E protein gene were mixed with adenoviral destination vector, pAd/CMV/V5-DEST (Invitrogen), to generate recombinant adenoviral plasmid containing E protein gene. After transforming the recombinant adenoviral plasmid DNA into competent E. coli, we extracted and purified DNA from selected putative positive clones identified by PCR amplification and gel electrophoresis. Those putative clones were also cultured on LB plates containing 30 µg/ml chloramphenicol, since true expression clones would be ampicillin-resistant and chloramphenicol- sensitive. Following digestion of the recombinant adenoviral plasmid DNA containing E protein gene with the restriction enzyme PacI, human embryonic kidney 293A cells were transfected to generate replication-incompetent adenovirus. Culture medium was replaced with fresh complete culture medium every 2~3 days until visible regions of the cytopathic effect (CPE) were observed. When approximately 50~70% CPE was observed, adenovirus-containing cells and media were harvested. The expression of E protein was identified with RT-PCR after NIH3T3 cells were infected with rAd-E. The replication-incompetent adenovirus expressing E protein of DenV2 (rAd-E) were purified with the Adeno-X mini purification kit (Clontech, Mountain View, CA), titrated by plaque assay, and stored at -80℃ until use.
Construction of vaccinia virus expressing E protein
Recombinant vaccinia virus expressing E protein of DenV2 (VV-E) was constructed using the shuttle vector, pSC11 (provided by Dr. B. Moss, NIH, Bethesda, MD) (37). Plasmid pSC11 encoding E protein gene was subcloned from pGEMT/E vector by restricting the plasmid using BglII and Sca I endonucleases. Human thymidine kinase-deficient 143B cell (HuTK-cells) were grown to 80% confluency in Eagle's Minimum Essential Medium (EMEM) containing 5% FBS. They were infected with vaccinia virus strain WR at a multiplicity of infection (MOI) of 0.05 followed by transfection with pSC11 encoding E protein gene using Lipofectamine (Gibco-BRL, Grand Island, NY). After CPE had developed (usually in 48~72 h), the cells were ruptured and the cell lysates containing the putative recombinant virions were employed to plaque isolation in the presence of bromodeoxyuridine (BrdU; 25 µg/ml) and Bluo-gal (0.6 mg/ml; Gibco-BRL). Blue plaques, produced by replicating recombinant virion expressing the lacZ gene, were collected and used to enhance the virus content of the plaques by inoculating a confluent layer of HuTK- cells. Recombinant vaccinia virus expressing E protein of DenV2 were harvested, plaques purified and enhanced by infecting larger volumes of two or more cell monolayers. The expression of E protein by recombinant vaccinia virus was confirmed by immunoblot. The titers of recombinant vaccinia virus were determined by plaque assay and stored at -80℃ until needed.
Protocol for vaccination and sample collection
Groups of mice (5- to 6-weeks old female) were immunized with either 100 µg of pDE or 106 PFU of rAd-E and VV-E via the intramuscular (i.m.) route. In prime-boost experiments, primarily immunized mice were boosted 7 days later with alternative vaccine vehicle via the same route. The i.m. immunization was performed by injecting the indicated immunogen into the anterior tibialis muscle. Control mice were given the empty vector (pCI-neo), replication-incompetent adenovirus expressing the LacZ gene (rAd-LacZ), and vaccinia virus expressing chicken ovalbumin (VV-OVA). Serum samples were collected by retro-orbital bleeding and stored at -80℃ until needed.
ELISA for E specific antibody, IgG, IgG1, and IgG2a
A standard enzyme-linked immunosorbent assay (ELISA) was used to determine the levels of E-specific antibodies in the serum samples. Briefly, ELISA plates were coated overnight at 4℃ with E protein (100 ng/well) in the sample wells and goat anti-mouse IgG/IgG1/IgG2a (Southern Biotechnology Associate Inc., Birmingham, AL) in standard wells. E protein of DenV2 was purified from recombinant E. coli by the Ni-NTA His-Tag column (Peptron Co. Ltd, Daejeon, Korea). The plates were washed three times with PBS-Tween 20 (PBST) and blocked with 2% non-fat dehydrated milk. The samples were serially diluted 2-fold, and incubated for 2 h at 37℃. This was followed by incubation with horseradish peroxidase-conjugated goat anti-mouse IgG/IgG1/IgG2a for 1 h. The color was developed by the addition of a suitable substrate (11 mg of 2,2-azinobis-3-ethylbenzothiazoline-6-sulfonic acid in a mixture of 25 ml of 0.1 M citric acid, 25 ml of 0.1 M sodium phosphate, and 10 µl of hydrogen peroxide). The concentration of E-specific antibodies was determined using an automated ELISA reader and the SOFTmax Pro4.3 program (Spectra MAX340: Molecular Device, Sunnyvale, CA).
Cytokine ELISA following in vitro stimulation of CD4+ T cells
To examine cytokine production from stimulated CD4+ T cells, splenocytes prepared from immunized mice were used as responder cells. The syngeneic antigen-presenting cell (APC) populations enriched by OptiPrep™ gradient (13.8% iodixanol; Axis-Shield, Oslo, Norway) (38) were pulsed with E protein (100 µg/ml) and subsequently used as stimulators. The responder and E protein-pulsed stimulator cells were combined at responder-to-stimulator ratios of 5:1, 2.5:1, and 1.25:1 in 200 µl of RPMI medium. The culture supernatants were harvested after 3 days of incubation. A similar number of responder cells were stimulated with 5 µg of concanavalin A for 48 h as a polyclonal positive stimulator.
ELISA was used to determine cytokine levels of IL-2, IL-4, and IFN-γ in the culture supernatants. Briefly, 100 ng per well of either IL-2, IL-4, or IFN-γ anti-mouse antibody (eBioscience, San Diego, CA; clone no. JES6-1A12, 11B11, and R4-6A2, respectively) was added to each ELISA plate. The plates were then incubated overnight at 4℃ and then washed three times with PBS-Tween 20. Next, they were blocked with 3% non-fat dried milk for 2 h at 37℃. The culture supernatant and recombinant IL-2, IL-4, and IFN-γ protein (Pharmingen, San Diego, CA) as standards were used. Each of these reagents was serially diluted two-fold, and then added to the corresponding plates. The plates were then incubated overnight at 4℃. Next, biotinylated IL-2, IL-4, and IFN-γ antibodies (eBioscience; clone no. JES6-5H4, BVD6-24G2, and XMG1.2, respectively) were added, after which the plates were incubated at 37℃ for an additional 2 h. The plates were then washed and incubated with peroxidase-conjugated streptavidin (Pharmingen) for 1 h, after which the color was developed with the addition of a substrate (2,2-azinobis-3-ethylbenzothiazoline- 6-sulfonic acid) solution. The concentrations of cytokines were then determined using an automated ELISA reader and the SOFTmax Pro4.3 program to compare the samples to two concentrations of standard cytokine protein. Data were expressed by subtracting the produced cytokine levels of no E protein-treated cultures from cytokine produced from E protein-stimulated cultures.
Intracellular CD154 staining for E-specific CD4+ T cells
We used intracellular CD154 staining to identify E protein-specific CD4+ T cells, as previously described (39,40). Briefly, 106 freshly explanted splenocytes per well were stimulated with E protein (100 µg/ml) in U-bottom 96-well plates for 12 h. Brefeldin A (2 µg/ml) was added for the last 6 h culture period to facilitate intracellular CD154 accumulation. After 12 h-stimulation, cells were washed twice with PBS containing 1% BSA, 0.05% NaN3, and 2 µg/ml brefeldin A. Cells were subsequently incubated with FITC-conjugated anti-CD4 for surface staining, followed by fixation with PBS containing 10% formaldehyde. The surface-stained cells were then permeabilized, washed, and stained intracellularly by incubation with PE-conjugated anti-CD154 for 30 min at room temperature. After several washes, the intracellular CD154 molecules were determined by flow cytometry.
In vivo CTL killing activity
An in vivo CTL assay was conducted as reported elsewhere (41). Splenocytes were collected from recipient mice 24 h after adoptive transfer of target cells that were previously pulsed with E331-339 (H-2Ld; SPCKIPFEI) eptiope peptide (1 µg/ml) and labeled with CFSE (2.5 µM), and then analyzed by flow cytometry. To control for antigen specificity, unpulsed syngeneic splenocytes previously labeled with CFSE (0.25 µM) were injected intravenously (i.v.) along with target cells. Each population was distinguished by their respective fluorescence intensity. The percentage of killing of target cells in immunized mice was calculated using the following equation: ratio = (percentage of CFSElow/percentage of CFSEhigh). The percentage of specific lysis = (1 - [ratio of naïve/ratio of immunized])/100.
Intracellular IFN-γ staining for E331-339-specific CD8+ T cells
Single-cell suspension (5×106 cells/ml) of splenocytes isolated from immunized mice was incubated with E331-339 (H-2Ld; SPCKIPFEI) epitope peptide (2 µg/ml) in RPMI media supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin at 37℃. After 6 h-incubation, the stimulated splenocytes were employed to stain surface CD8 followed by staining intracellular IFN-γ. The IFN-γ-producing CD8+ T cells was determined by flow cytometry.
RESULTS
Humoral immune responses induced by plasmid DNA and live viral vectors expressing E protein
To evaluate the immune responses induced by plasmid DNA (pDE), recombinant adenovirus (rAd-E) and vaccinia virus (VV-E) expressing E protein of DenV2, groups of mice were immunized with these constructs and the levels of E-specific IgG in sera were determined 10 days post-immunization. pDE was observed to induce lower levels of E-specific IgG, when compared to live viral vectors (Fig. 1A). VV-E induced significantly higher levels of E-specific IgG than rAd-E at the equivalent amount of immunogens (106 PFU/mouse). When the distribution of E-specific IgG isotypes, IgG1 and IgG2a, was evaluated, VV-E showed significantly higher levels of E-specific IgG1 and IgG2a, compared to pDE and rAd-E (Fig. 1B and C). However, three constructs expressing E protein of DenV2 showed comparable ratios of IgG2a to IgG1 each other (Fig. 1D). These results indicate that constructed pDE, rAd-E and VV-E could successfully induce humoral responses specific for E protein of DenV2.
CD4+ T cell responses induced by plasmid DNA and live viral vectors expressing E protein
After immunizing mice with pDE, rAd-E and VV-E, the cytokine production from CD4+ T cells in response to E protein stimulation was determined 14 days post-immunization. CD4+ T cells prepared from VV-E immunized mice produced higher amounts of IL-2 and IFN-γ in response to E protein stimulation, compared to those isolated from mice immunized by either pDE or rAd-E (Fig. 2 A~C). In contrast, immunization with rAd-E exhibited a significantly higher production of IL-4 compared to other forms of immunization (Fig. 2B). To understand immune responses induced by pDE, rAd-E or VV-E in detail, we determined the number of CD4+ T cells specific for E protein using intracellular CD154 staining (39,40). In consistent, VV-E immunization showed higher percentages of CD154+CD4+ T cells in response to E protein stimulation (Fig. 2D). Therefore, this result indicates that plasmid DNA and live viral vectors expressing E protein induced CD4+ T cell responses and VV-E immunization induced the most potent immune responses.
CD8+ T cell responses induced by plasmid DNA and live viral vectors expressing E protein
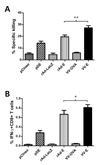
To better understand immune responses induced by plasmid DNA, live viral vectors expressing E protein of DenV2, we examined the in vivo CTL killing activity specific for E331-339 (SPCKIPFEI) epitope. Similarly, immunization with VV-E showed the most potent activity of in vivo CTL, compared to other types of immunization (Fig. 3A). When the percentage of IFN-γ-producing CD8+ T cells in response to E331-339 (SPCKIPFEI) epitope stimulation was determined, mice immunized with VV-E displayed a higher percentage of IFN-γ-producing CD8+ T cells than others (Fig. 3B). In line with antibody and CD4+ T cell responses, this result indicates that VV-E immunization could induce more potent immune responses than pDE and rAd-E.
Humoral immune responses induced by alternating prime-boost immunization using plasmid DNA and live viral vectors expressing E protein
To compare E-specific immune responses induced by alternating prime-boost immunization with pDE, rAd-E, and VV-E, we first assessed the humoral responses following immunization with the indicated protocols (Fig. 4A). The primary immunization with vaccinia virus expressing E protein followed by boosting with adenovirus expressing E protein showed highest levels of E-specific IgG, but E-specific IgG lasted for 14 days post-boosting with higher levels in the group that received priming with rAd-E and boosting with VV-E. In general, it was observed that the levels of E-specific IgG in sera rapidly decreased when VV-E was used as immunogen of primary vaccination. Furthermore, priming with VV-E and boosting with rAd-E led to production of E-specific IgG1 isotype with higher levels than the other group, when the levels of E-specific IgG isotypes, IgG1 and IgG2a, were determined (Fig. 4B and C). Notably, if VV-E was used for primary vaccination, the levels of E-specific IgG1 isotype were maintained till 14 days post-boosting, but E-specific IgG2a levels decreased. Therefore, these results suggest that E-specific IgG and its isotypes, IgG1 and IgG2a, may be produced at various levels by alternating prime-boost immunization using DNA vaccine and live viral vector, depending on the properties of immunogens used for primary vaccination.
CD4+ T cell responses induced by alternating prime-boost immunization using plasmid DNA and live viral vectors expressing E protein
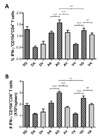
When the production of Th1- and Th2-type cytokines by CD4+ T cells stimulated with E protein was analyzed, the results showed a different pattern from E-specific IgG (Fig. 5). The group of mice that received priming with rAd-E and boosting with pDE showed a higher production of IL-2 and IFN-γ from stimulated CD4+ T cells (Fig. 5A and C). However, priming with VV-E and boosting with rAd-E that showed the highest level of E-specific IgG induced lower production of IL-2 and IFN-γ from stimulated CD4+ T cells. IL-4 production by CD4+ T cells showed no pattern correlated with the protocols of alternating prime-boost vaccination using DNA vaccine and live viral vectors (Fig. 5B). Furthermore, when E-specific CD4+ T cells producing IFN-γ were enumerated by intracellular CD154 and IFN-γ staining (39,40), the results revealed that priming with rAd and boosting with pDE induced the highest number of IFN-γ-producing E-specific CD4+ T cells than other immunization (Fig. 6A). Similarly, groups of mice that received priming with rAd-E and boosting with VV-E exhibited a higher number of IFN-γ-producing E-specific CD4+ T cells in spleen (Fig. 6B). Of particular interest, priming with pDE showed inhibited CD4+ T cell responses after boosting with either rAD-E or VV-E. Taken together, these results indicate that CD4+ T cell responses achieved by priming with adenovirus expressing E protein and boosting with DNA vaccine were superior to other immunization protocols, and primary responses induced by DNA vaccine may interfere with CD4+ T cell responses of liver viral vector as booster.
CD8+ T cell responses induced by alternating prime-boost immunization using plasmid DNA and live viral vectors expressing E protein
When CD8+ T cell responses specific for E protein of DenV2 were determined by in vivo CTL killing activity, the results revealed that priming with VV-E and boosting with pDE showed the most potent in vivo CTL killing activity (Fig. 7A). This exceeded the results achieved by priming and boosting with the same vaccine VV-E, and was superior to responses induced by priming with rAD-E and boosting with pDE. Groups of mice that received pDE as primary immunogen showed lower in vivo CTL killing activity than with other methods. Consistently, priming with VV-E and boosting with pDE showed a higher number of IFN-γ-producing CD8+ T cells in response to stimulation with E331-339 (SPCKIPFEI) epitope peptide (Fig 7B and C). Also, the number of IFN-γ-producing CD8+ T cells induced by primary immunization with pDE was not increased by boosting with live viral vectors including rAd-E and VV-E. These results indicate that boosting with DNA vaccine expressing E protein of DenV2 following primary immunization with live viral vectors elicits strong CD8+ T cell responses.
DISCUSSION
We demonstrate that DNA vaccine, adenovirus, and vaccinia virus expressing E protein of DenV2 induced differential immune responses. Among three constructs, VV-E induced the most potent IgG responses, Th1-type cytokine production by stimulated CD4+ T cells, and CD8+ T cell response. rAd-E showed higher production of Th2-type cytokine IL-4 compared to other constructs, DNA vaccine and vaccinia virus. Furthermore, when such constructs were used for alternating prime-boost vaccination, the results revealed a different pattern of immune responses, depending on the constructed vaccine used for the priming step. Notably, priming with VV-E induced higher E-specific IgG levels which rapidly decreased compared to the group that received rAd-E as a priming vaccine. Also, strong CD8+ T cell responses specific for E protein were induced when VV-E was used for priming the vaccine vehicle, and such CD8+ T cell responses were significantly boosted with pDE vaccination, compared to booster vaccination with either rAd-E or VV-E. In contrast, priming with rAd-E induced stronger CD4+ T cell responses which subsequently boosted DNA vaccination more than others, as determined by intracellular CD154 and IFN-γ staining. Therefore, these results indicate that priming with live viral vector vaccine could lead to a different pattern of E protein- specific immune responses that were significantly more enhanced by booster vaccination with DNA vaccine, compared to live viral vector vaccine.
Vaccination protocols commonly require multiple immunizations to achieve robust, protective, and sustained immune responses. In particular, heterologous prime-boost vaccination with DNA vaccine and live viral vector vaccine has emerged as an effective strategy for eliciting a robust response to target antigen (25,26). In such vaccination strategies, the most effective approach has proven to be priming with DNA vaccine and boosting with recombinant viral vector expressing the same antigen (25,26). This approach has been used extensively in the development of vaccines against a number of pathogens including human immunodeficiency virus (HIV) (42,43), herpesvirus (44), hepatitis C virus (HCV) (30), Ebola virus (45), and highly pathogenic avian influenza virus (HPAI) H5N1 (46,47). However, this dogma has sometimes been challenged by different results which show that priming with either live viral vector or live attenuated vaccine and boosting with DNA vaccine showed stronger responses than priming with DNA vaccine and boosting with live viral vector (27,28). In addition, priming with live viral vector vaccine and boosting with DNA vaccine induced the most potent immune responses at both systemic and mucosal sites if prime-boost vaccination is applied to mucosal route (48). Our results support the later notion at least in prime-boost vaccination against E protein of dengue virus type 2. Of particular interest, a different magnitude of CD4+ and CD8+ T cell responses were induced if either rAd-E or VV-E is used as priming vaccine vector. Minutely, priming with rAd-E induced higher number of CD4+ T cells specific for E protein following boosting with pDE, whereas stronger CD8+ T cell responses were induced by priming with VV-E followed by boosting with pDE. The reason that priming with viral vector vaccine and boosting with pDE showed effective immune responses may be explained by escape of DNA vaccine to interference conferred by pre-existing immunity (22,23,49). Conceivably, it is possible that pre-existing immunity induced by priming with pDE or live viral vector vaccine (rAD-E and VV-E) could inhibit immune responses provided by boosting with live viral vaccine expressing the same antigen, E protein. Indeed, the group that received pDE at both priming and boosting showed stronger CD4+ responses than the group that received priming with pDE and boosting with either rAD-E or VV-E (Fig. 6). However, how priming with either rAd-E or VV-E induced different patterns of CD4+ and CD8+ T cell responses following boosting with pDE remains to be explained.
Alternating prime-boost immunization is known to confer synergistically stronger responses to antigens and greater protection than immunization with either vaccine alone (25,26). However, the immunological basis for this outcome remains to be resolved. It is likely that the success of this approach may depend on several factors. In some instances, immune responses to repeated administration of the vector used for the primary immunization can neutralize the immunity induced by a booster, thereby inhibiting the effective immune responses that follow boosting. Also, the prime-boost vaccination may enable encoded antigen to be presented through alternative modes, depending on the diversity of antigen delivery. In particular, boosting with live viral vector may selectively expand the small population of antigen-specific CD4/CD8 T cells by inducing type I IFN production, leading to IL-15 production, which has been known to maintain CD8+ T cell proliferation and survival (50,51). Moreover, adenovirus appears to infect early dendritic cells (DCs), which may differentiate to mature DCs that present antigens more effectively (30,31). Adenovirus also synthesizes larger quantities of proteins that are taken up by endocytosis. Similarly, vaccinia virus can replicate in several types of epithelial cells as well as antigen-presenting cells to provide antigen for cognate lymphocytes. In the present study, we used replication-incompetent adenovirus that can provide antigen protein from infected cells, whereas vaccinia virus can replicate in some cells of immunocompromised hosts. Thus, it is possible that vaccinia virus could induce stronger responses compared to the adenovirus at equivalent antigen doses (Fig. 1). Also, the divergent cell targeting and antigen presentation of adenovirus and vaccinia virus complement each other in prime-boost vaccination, allowing a greater outcome of immune responses than with either vaccine vehicle alone.
Optimal vaccination to provide protective immunity against dengue virus is still challenging. One of the major issues to develop vaccines against dengue infection is how to effectively provide protective immunity against all four serotypes of the dengue virus. We hope that the alternating prime-boost vaccination using DNA vaccine, adenovirus, and vaccinia virus expressing E protein can be used as a effective prophylactic strategy to control fatal DHF and DSS caused by dengue infection. To achieve the practical development of vaccination strategies against dengue virus, the prime-boost vaccination using DNA vaccine and/or live viral vector vaccine expressing tetravalent antigen is recommended. Therefore, our observation will provide valuable information for the establishment of optimal prime-boost vaccination against dengue virus.





 XML Download
XML Download