

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Rheumatoid arthritis (RA) is characterized by a chronic inflammatory process, which primarily involves the synovial membrane and is typically associated with serologic evidence of systemic autoimmunity, culminating in loss of joint function (1). The presence of auto-antibodies, such as rheumatoid factor (RF) and anti-cyclic citrullinated peptide (anti-CCP) antibodies, can precede the clinical manifestations of RA by many years (2). Therefore, it is recommended that effective therapy be initiated as soon as autoimmune abnormalities appear (3); however, there are many unanswered questions regarding the pre-clinical stages of RA. The central questions relate to the factors that serve to initiate, amplify, and mature the immune responses in RA. It is difficult to investigate the molecular features in patients with RA before the onset of clinical symptoms, which is possible in animal models.
Collagen-induced arthritis (CIA) has been used to study the basic mechanisms of autoimmune arthritis because of the clinical, immunologic, histologic, and genetic similarity to RA (4-6). In this model, native type-II collagen (CII), when presented with an adjuvant, predictably and reproducibly precipitates a chronic inflammatory arthritis in the joints of genetically-susceptible animals. The development of CIA is thought to depend on T cells, and disease susceptibility is linked to the major histocompatibility complex (MHC) (7). CII-specific CD4+T cells have been reported to be fundamental in the initiation and perpetuation of CIA (8,9). Recently, abatacept, which targets T-cell modulation via the co-stimulatory CD80/CD86:CD28 pathway, exemplifies the rationale and efficacy of using T-cell modulation as a therapeutic approach (10). Therefore, the abnormality in T cell-related immunity plays a role in the pathogenesis of RA. In addition, the primary mechanism leading to tolerance to self-antigens is the deletion of self-reactive T cells in the thymus through the mechanism by which T cells with a high affinity for self-peptides undergo the active induction of apoptosis. Autoimmune arthritis in various animal models can be attributed to a defect in T cell selection within the thymus (11-13). Since, we questioned where and when T cell immunity dysregulation occurs, we investigated the gene expression of T cell-related molecules, in the thymus, peripheral blood and synovium during the initiation phase of CIA using cDNA microarray technology followed by validation using real-time PCR.
Go to : 
MATERIALS AND METHODS
Animals
Adult female Lewis rats (7 weeks old) weighing approximately 150 g were obtained from Harlan (Indianapolis, IN, USA), housed under standard conditions, and given free access to food and water. The rats were adapted to the new conditions for at least 7 days before the initiation of the studies. All experiments were conducted in accordance with the animal care guidelines of the National Institutes of Health and the Korean Academy of Medical Sciences. This study was approved by the Eulji University Animal Institutional Review Board.
Rat CIA model
Chicken CII (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in 0.05 M acetic acid to a final concentration of 4 mg/ml. CII emulsified in incomplete Freund's adjuvant (IFA) was homogenized at a 1:1 ratio using a PowerGen 35 (Fisher Scientific, Pittsburgh, PA, USA). For the induction of CIA, rats, 8 weeks old (n=3 per group), were immunized intradermally at the base of the tail with 0.2 ml of the emulsion on day 0, followed by one-half volume of each on day 7. Another group of rats were immunized only with and equivalent volume of IFA, only with CII on the same days. Non-immunized rats were used of control group.
Arthritis was assessed daily, and inflammation was scored, as described by Nanakumar and co-workers (14). In our previous study, the induction ratio of CIA was >86%.
Each experimental condition was reproduced three times (RNA isolation, probe preparation, and independent hybridization), and each replicate contained RNA samples pooled from three animals.
For gene expression analysis, the sample was collected on days 0 (control), 4, and 9. Sample tissues from three rats were collected at each time point. Blood was collected through the postcaval vein from each animal. Synovium was collected from the posterior knee joint. Samples were treated with RNAlater (Ambion, Austin, TX, USA) to prevent RNA degradation.
RNA preparation
Total RNA was extracted using Trizol (Invitrogen Life Technologies, Carlsbad, CA, USA) and purified using RNeasy columns (Qiagen, Valencia, CA, USA), according to the manufacturer's protocol. After processing with DNase digestion and clean-up procedures, the RNA samples were quantified, aliquotted, and stored at -80℃ until use. For quality control, RNA purity and integrity were evaluated by denaturing gel electrophoresis, the OD 260/280 ratio, and analyzed on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA).
Labeling and purification
Total RNA was amplified and purified using the Ambion Illumina RNA amplification kit (Ambion) to yield biotinylated cRNA, according to the manufacturer's instructions. Briefly, 550 ng of total RNA was reverse-transcribed to cDNA using a T7 oligo (dT) primer. Second-strand cDNA was synthesized, in vitro-transcribed, and labeled with biotin-NTP. After purification, the cRNA was quantified using a ND-1000 spectrophotometer (NanoDrop, Wilmington, DE, USA).
Hybridization and data export
Seven hundred fifty nanograms of labeled cRNA samples were hybridized to each Rat-12 expression bead array for 16~18 h at 58℃, according to the manufacturer's instructions (Illumina Inc., San Diego, CA, USA). Detection of array signals was carried out using Amersham fluorolink streptavidin-Cy3 (GE Healthcare Bio-Sciences, Little Chalfont, UK), according to the bead array manual. Arrays were scanned with an Illumina bead array Reader confocal scanner, according to the manufacturer's instructions. Array data export processing and analysis was performed using Illumina BeadStudio.
Raw data preparation and statistical analysis
The quality of hybridization and overall chip performance were monitored by visual inspection of internal quality control checks and the raw scanned data. Raw data were extracted using the software provided by the manufacturer (BeadStudio v. 3.3.0). Array data were filtered by a detection p-value<0.05 (similar to signal-to-noise) in at least 50% of the samples. We applied a filtering criterion for data analysis; a higher signal value was required to obtain a detection p-value<0.05. The selected gene signal value was transformed by logarithm and normalized by the quantile method. Comparative analyses were carried out using a |fold|>2. Hierarchical cluster analysis was performed using complete linkage and Euclidean distance as a measure of similarity. All data analyses and visualization of differentially-expressed genes were conducted using ArrayAssist® (Stratagene, La Jolla, CA, USA). Ontology-based analyses were performed using the Panther database (http://www.pantherdb.org).
Reverse transcription and real-time PCR
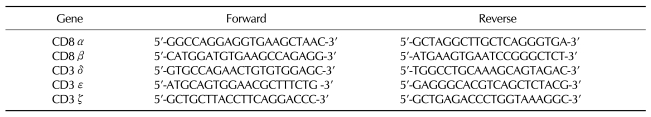
Reverse transcription (RT) was carried out according to the instructions in the ImProm-II™ reverse transcription system (Promega, Madison, WI, USA). In brief, 1 µg of total RNA was reverse-transcribed in a total volume of 20 µl using an oligo-dT primer. The RT reaction was performed at 25℃ for 5 min, 42℃ for 1 hr, and 70℃ for 15 min. One µl of cDNA was used as a template for real-time PCR in a 25 µl reaction mixture, including 0.75 U of GoTaq DNA polymerase (Promega), 5 µl of 5X buffer, 0.2 mm of dNTP (Promega), 2.5 µl of 3000X SYBR Green I (BMA, Rockland, ME, USA), and 10 pM of each primer. Oligonucleotide primers for real-time PCR were prepared using Primer 3 software (Whitehead Institute/MIT Center for Genomes Research, Cambridge, MA; http://www.bioneer.co.kr/cgi-bin/primer/primer3.cgi). Information and sequences of primers of the steroidogenic enzymes tested in the present study are summarized in Table I. The PCR program used an initial step of 95℃ for 5 min for pre-denaturation, followed by denaturation at 94℃, annealing, and extension at 72℃ using the PTC-200 Chromo 4 real-time system (Bio-Rad Laboratories, Hercules, CA, USA). The final extension was carried out for 10 min at 72℃. Beta-actin was included as an internal PCR control. For quantification of real-time PCR results, the relative standard curve method was used to obtain quantitative values. Each sample was replicated three-to-four times, and the normalized mean value to beta-actin was used for final comparison.
Real-time PCR data presentation and statistical analysis
Data for mRNA abundance were expressed relative to the control and CIA groups on days 4 and 9. In the figures, data are presented as the mean±standard error. Comparisons between CIA and controls were performed using one-way analysis of variance, followed by the Mann-Whitney U test, using SPSS software (SPSS Inc., Chicago, IL, USA). In all cases, a p<0.05 was considered statistically significant.
Histology
At the end of each experiment, the knee joints were fixed in 4% formalin for 2 days, decalcified in EDTA for 3 weeks, then embedded in paraffin. From the paraffin-embedded tissue blocks, 2-µm sections were cut and stained with haematoxylin-eosin for histologic analysis.
Go to :
RESULTS
A great number of genes were significantly modulated during the initiation stage of CIA
To define the gene expression profile in the initiation stages of CIA, we used a expression chip with 21,910 gene specificities. First, gene expression was compared between the CIA and control groups on days 4 and 9 after the 1st immunization. We observed 1,243 genes were expressed differentially in CIA on day 4 than on day 9, which could be attributable to a biological process or molecular function (Fig. 1). Table II shows the list of up- or down-regulated genes according to the organ in which they were differentially-expressed. We found that the synovium was the organ in which most genes were differentially-expressed, and only one gene, Il1f8, was differentially-expressed in the blood. No gene was differentially-expressed in the thymus.
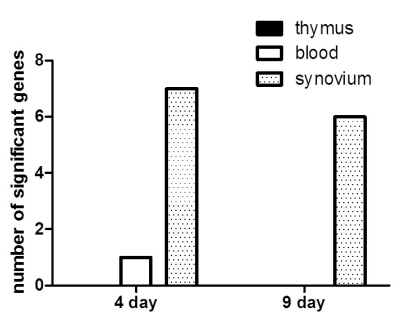 | Figure 1Number of immune-related genes differentially-expressed in the early phase of CIA. RNA samples were collected from the thymus, blood, and synovium of the CIA and control groups on days 4 and 9 after the 1st immunization. A RatRef-12 expression beadchip with 21,910 gene specificities was used to define the gene expression profile in CIA and control rats. Only immune-related genes differentially-expressed between the CIA and control groups are shown here. Most genes differentially-expressed in CIA were shown in the synovium on days 4 and 9 (dotted bar), whereas 1 gene in the blood was shown on day 4 (white bar), and no genes were shown in the thymus (black bar).
|
Differential expression of genes encoding T-cell receptors (TCRs) in arthritic synovial tissue of CIA
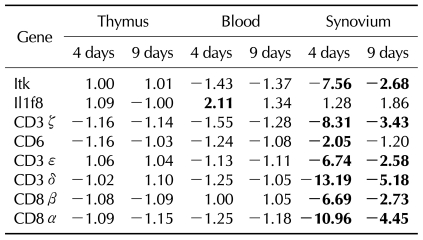
Since we founded on T cell associated genes, we analyzed genes related to TCR/CD3 complex, including CD3δ, CD3ε, and CD3ζ. Interestingly, most genes encoding TCR were down-regulated in CIA (Fig. 2A). Day 4 showed a greater difference in gene expression than day 9 between CIA and controls. Next, we analyzed which organ showed differentially expressed genes. The synovium was the only organ in which the genes were differentially expressed between CIA and control groups, whereas no difference was found in the thymus and blood.
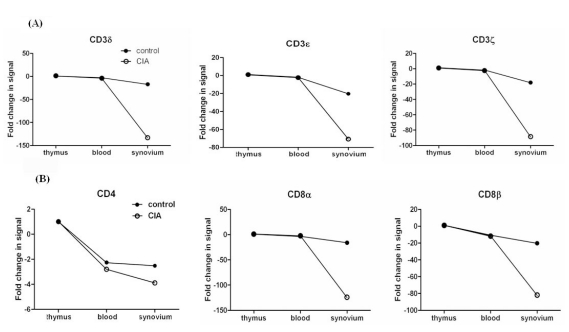 | Figure 2The ratio of TCR-related gene expression of the CIA-to-control group in each organ in the initiation phase of CIA. TCR-related gene expression was compared between the CIA and control groups. The data shown here are the ratio of gene expression of the CIA-to-control group in each organ. (A) CD3 complex gene expression. CD3δ, CD3ε, and CD3ζ gene expression was down-regulated in the synovium of CIA, with no differences in the thymus and blood. (B) CD4 and CD8 gene expression. CD8α and CD8β gene expression was down-regulated in the synovium of CIA, with no difference in the thymus and blood. No difference in CD4 gene expression existed between the CIA and control groups in the synovium, thymus, or blood.
|
Next, we analyzed gene expressions of CD4 and CD8 which are another T cell surface molecules that play an additional role in T cell recognition (15). The expression of the CD8α and CD8β genes were reduced significantly in CIA, howerer the CD4 gene was expressed similarly between CIA and control groups (Fig. 2B). To validate our microarray findings, we performed real-time RT-PCR on the genes of TCR/CD3 and T cell co-receptors using independent animals. The expression of CD3δ, CD3ε, CD3ζ, CD8α, and CD8β, as revealed by RT-PCR, was in agreement with the microarray data (Fig. 3).
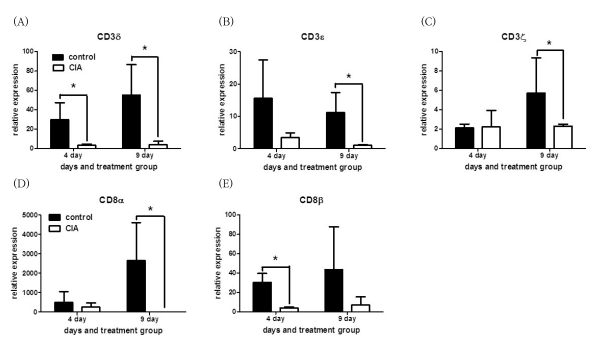 | Figure 3Validation of the CD3 complex and CD8 gene expression in CIA and control groups. RT-PCR was performed using SYBR and a specific primer set for (A) CD3δ, (B) CD3ε, (C) CD3ζ, (D) CD8α, and (E) CD8β. The RNA samples are independent of the samples used for microarray analysis. Data are expressed as the mean±SD. *p<0.05 by the Mann-Whitney U test.
|
Gene expression profile in rats immunized only with CII or adjuvant
We next determined whether the differential expression of T cell-mediated genes was more affected by CII or adjuvant. We compared four groups of rats, including rats immunized both with IFA and CII (CIA), rats immunized only with IFA (rIFA), rats immunized only with CII (rCII), and unimmunized rats. Asshown in the previous analysis between the CIA and control groups, gene expression profile showed greater difference on day 4 than day 9 both in rCII and rIFA (Fig. 4). Most genes were differentially expressed in the synovium, whereas the thymus showed only one gene differentially-expressed in rCII on day 9.
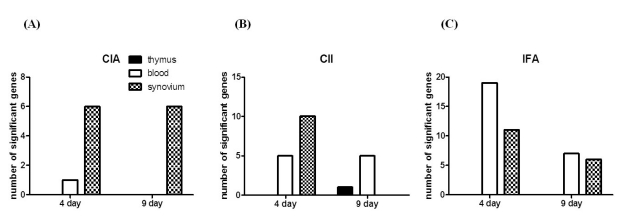 | Figure 4Number of genes differentially-expressed in CIA and rats immunized only with CII or adjuvant. To define the gene expression profile in the initiation stages of CIA, we used a RatRef-12 expression beadchip (Illumina Inc., San Diego, CA, USA) with 21,910 gene specificities, and compared the following 3 groups of rats with the control group: (A) IFA/CII immunized (CIA), (B) CII immunized, and (C) IFA (MDP). Samples were collected from the thymus (black bar), blood (white bar), and synovium (dotted bar).
|
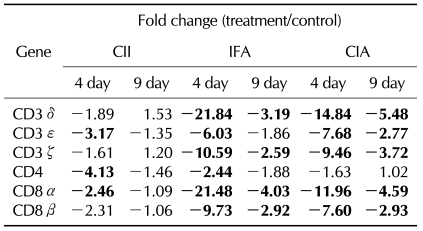
Interestingly, the synovium of CII showed down-regulation of one CD3 complex gene, CD3ε, whereas the synovium of IFA showed down-regulation of more genes including CD3δ, CD3ε, and CD3ζ on day 4, as shown in CIA (Table III). Furthermore, the difference of gene expression compared to controls was greater in rIFA than in CIA. In terms of T cell co-receptor, even though both the synovium of rCII and rIFA showed down-regulation of CD8α and CD8β, a greater difference in gene expression was shown in rIFA compared to rCII or CIA. CD4 gene expression was decreased in rIFA compared to rCII, which was not shown in CIA.
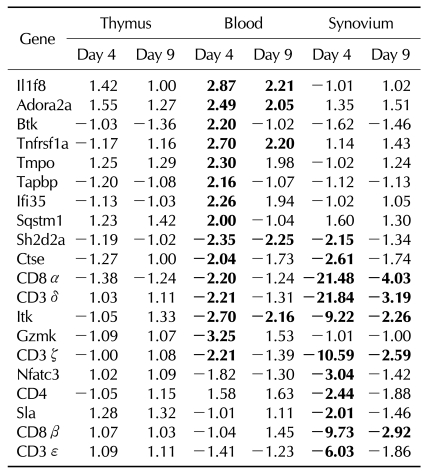
In addition, other genes were expressed differentially on rCII or rIFA, which was not differentially expressed in CIA. The blood of rCII revealed up-regulation of Btk, Tmpo, and Tapbp on day 4, and up-regulation of IL9r and Ciita, and down-regulation of Ctss and Sh2d2a on day 9. The synovium of rCII showed up-regulation of Sqstm1 and down-regulation of Tapbp, Hla-dma, and Nfatc3 (Table IV).
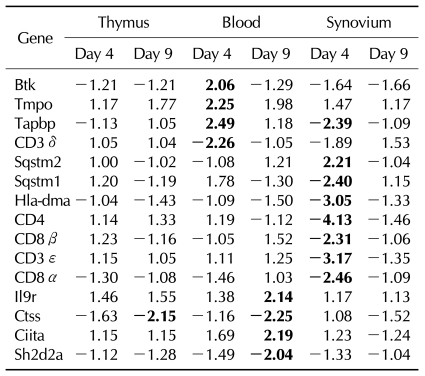
The blood of rIFA showed up-regulation of IL1f8, Adora2a, Tnfrsf1a, and Ifi35, in addition to that showed the genes that showed increased expression in rCII on day 4. The Sh2d2a, Ctse, Gzmk, and Itk were down regulated in the blood of rIFA, and the synovium of rIFA showed down-regulation of Sh2d2a, Ctse, Nfatc3, Sla, and Itk on day 4 (Table V).
Pathologic analysis of synovium in CIA rats and controls
We also examined synovia pathologically at the initiation time, but there were no differences among the four groups, including CIA, rIFA, rCII, and unimmunized. These results indicated that there were changes at the molecular level before histologic changes began, including synovial hypertrophy, pannus formation, cartilage destruction, and bone erosion.
Go to :
DISCUSSION
During the 1990s, the widely accepted concept of cytokine networks perpetuating disease, with tumor necrosis factor (TNF)-α assuming a hierarchical role (16), was supported by the success of TNF-α-blocking treatment in RA (17,18). However, the lack of universal benefit from TNF-α-blocking therapy (19) has focused attention to alternative upstream immunologic mediators. The central questions relate to the factors that serve to initiate, amplify, and mature the immune responses in RA. Gene expression profiles at different stages of CIA have been reported previously (20). However, it is difficult to determine the causal genes in those studies because too many genes were already expressed when inflammatory features developed. Thus, we were interested in gene expression profiles at an earlier time point, such as immediately after the 1st immunization. Especially, we investigated focused on T cell immunity-related genes because several observations over the years supported a fundamental role for T cells in RA (21,22). RA synovium contains a rich infiltrate of activated CD4+ cells and MHC class II molecules (23) with a correlation between the number of synovial T cells and the severity of joint damage (24). In addition, we considered which organs would be involved in dysregulation of T cell-mediated immunity in RA, because there were several lines of evidence that thymic abnormalities were associated with the pathogenesis of RA. The identification of other joint-specific antigens, such as gp-39 in thymic Hassall's corpuscles and medullary epithelial cells, provide new insight into the mechanisms of RA pathogenesis and may lead to more specific and physiologic methods of immune-modulation (25). Therefore, we investigated central immune organ, even thought thymus would not be affected by immunization. We determined the genome-wide expression profile of the thymus in addition to synovial joints and peripheral blood.
Our original hypothesis was that T cells could escape negative selection because intracellular signal transduction of thymocytes was dysregulated. The TCR/CD3 complex is one of the most intricate membrane receptor structures because it consists of six distinct chains (26,27). The clonotypic α and β chains of the TCR are responsible for recognizing antigen embedded in the MHC molecule present on the surface of antigen-presenting cells (APCs). The remaining chains, collectively called the CD3 complex, include the CD3-γ, -δ, -ε, and -ζ chains (26). T cells implicated in chronic inflammatory diseases, such as RA, respond weakly when stimulated in vitro with mitogen or antigen (28). Decreased expression of the TCR-associated CD3ζ chain has been suggested, but the mechanism underlying this hypo-responsiveness is unclear (29-33). Therefore, we investigated when TCR started to be decreased in the course of CIA development. We focused on the initiation time before looking at the whole picture through the total period of CIA development.
In our study, TCR down-regulation occurred at an early phase of CIA, and most genes expressed differentially were found in the synovium, not in the thymus and blood. Synovium tissue contains heterogeneous population of stromal cells and immune cells. Especially, there was definitely higher number of lymphocyte infiltrated in the synovium of CIA than control. Therefore, the expression of T cell receptors should be increase in CIA. However, we found the opposite result which is consistent with the result of former studies in which TCR-associated CD3ζ chain is decreased in CIA and RA (31,33). It was leading to the hypothesis that the synovium might play a pathologic role on T cells which remains to be clarified.
It has been reported that there is a correlation between the levels of TNF-α produced after anti-CD3 or PHA stimulation and the expression of CD3ζ (34). However, in our study, TNF-α had not been expressed until day 9. Therefore, we could guess that TNF-α was not involved in CD3ζ down-regulation, which remains to be clarified.
Another T cell surface molecules, CD4 and CD8, play a critical role in T cell recognition and activation by binding to their respective class II and I MHC ligands on APCs. In addition, CD4 and CD8 are also involved in post-binding events that lead to CTL activation and subsequent lysis of the target cells (35).
CD8 T cells are part of the T cell pool infiltrating the synovium in RA. However, the role of CD8 T cells in the pathogenesis of RA has not been fully delineated. In our study, the gene expression of CD8 receptors also decreased in the CIA group compared to the control group. Therefore, with down-regulation of the CD3 complex and CD8 receptors, T cells would be severely defective in activation.
Next, we questioned which factor would play a greater role in the down-regulation of TCR genes. The synovium from the rats immunized only with CII showed down-regulation of only one gene, CD3ε, whereas the synovium in the rats immunized only with adjuvant showed down-regulation of CD3δ, CD3ε, and CD3ζ, which showed a greater difference of gene expression compared to CII-immunized rats or CIA. T cell co-receptors, CD8α, and CD8β, which were down-regulated in CIA, showed a greater difference of gene expression in adjuvant-immunized rats compared to CII-immunized rats or CIA. CD4 gene expression was decreased both in adjuvant-immunized rats and CII-immunized rats, which was not shown in CIA. Taken together, adjuvant played a greater role in the down-regulation of the CD3 complex and co-receptors. Therefore, infectious triggers might play an important role in the down-regulation of the TCR gene CIA.
Furthermore, more genes were shown to be expressed differentially when we immunized rats with CII or adjuvant compared to the CIA. Even the blood in the rats immunized with CII only and with adjuvant only revealed another genes which were expressed differentially, including Btk, Tmpo, Tapbp, IL9r, Ciita, Ctss, Sh2d2a, Sqstm1, Tapbp, Hla-dma, Nfatc3, IL1f8, Adora2a, Tnfrsf1a, Ifi35, Ctse, Gzmk, and Itk. The function of those genes in the pathogenesis of CIA remains to be elucidated.
Go to :
 XML Download
XML Download