

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
NADPH oxidase (NOX) complex is composed of membrane-associated protein p22phox and the cytosolic proteins p47phox, p40phox, p67phox and Rac, a small GTPase and cloned in phagocytes (1). The first clone, phagocyte NOX (PHOX) is referred as NOX2 or gp91phox constitutively associates with p22phox. Subsequent association of p47phox, p40>phox, and p67phox to the NOX2/p22phox complex activates NOX2 function in the membrane and generates superoxide, resulting in promoted production of a large amount of reactive oxygen species (ROS) in the cytosol as well as in membrane (2,3). Although overproduction of ROS causes damages in DNA, lipids, and proteins (4), optimal production of ROS is required for the control of cell proliferation, apoptosis, hormone synthesis, and organogenesis (2,5). In particular, NOX2 complex is essential in innate immunity against microbial infections, as evidenced that mice lacking p47phox or gp91phox were highly susceptible to microbial infection, developed severe inflammation (6,7). Moreover, inherited abnormalities of NOX2, p22phox, p47phox or p67phox are closely associated with development of chronic granulomatous disease displaying insistent inflammation in lung, liver, and kidney (8,9). It is recently reported that NOX2-null mice develop spontaneous arthritis without antigenic stimulation, which are dependent of aging and favorable in females (10). Nox2 deficiency fails to generate regulatory T (Treg) cells, augmenting inflammatory cytokine production in T cells.
Sensitization and challenge of ovalbumin (OVA) antigen into the lung induces inflammation in the airways through activation of lung and peripheral immune cells and recruitment into the lung (11-13). A variety of immune cells such as macrophages, neutrophils, eosinophils, and mast cells are known to involve in the lung inflammation. Particularly, CD4+ T cells are highly responsible for inducing inflammatory immune response to antigenic stimulation (13). CD4+ T helper (Th) precursor cells are stimulated by T cell receptor activation and are then differentiated into the effector Th cells including Th1, Th2, and Th17 cells by coordination with different cytokine stimuli. Regulatory T (Treg) cells are also generated from Th precursor cells by addition of TGF-β inhibiting the inflammatory immune response. OVA injection prefers to develop effector Th2 and Th17 cells and increases the amounts of IL-4, IL-5, IL-13 and IL-17 cytokine in the lung, mainly contributing to the inflammatory response in the process of airway inflammation (14). Although cytokines in the inflamed tissue are critical regulators for determining immune responses, reactive oxygen species generated by NOX2 complex are recently suggested to be essential for controlling Th cell-mediated inflammation (15).
In this study, we investigated whether allergen-induced airway inflammation was stimulated in NOX2 deficiency. The absence of NOX2 significantly developed severe airway inflammation which was induced by ovalbumin injection, exhibiting increased red blood cell and immune cell infiltration in the lung, intensified mucus-secreting goblet cells, accumulated collagen deposition in the airway, and augmented inflammatory cytokine production.
MATERIALS AND METHODS
Materials
Anti-CD3 Ab was purchased from BD Pharmingen (San Jose, CA, USA) and cytokines and abs for ELISA were obtained from R&D Systems (Minneapolis, MN, USA). Ovalabumin (OVA, A5503) was purchased from Sigma-Aldrich Inc. (St Louis, CA, USA).
Mice
C57BL/6 wild type (WT) and NOX2 knockout (KO) mice (7) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and housed under specific pathogen-free conditions at Ewha Womans University. All animal experiments were performed in accordance with the guidelines of the institutional Animal Care and Use Committee (IACUC) and were approved by the IACUC committee at Ewha Womans University (ELAGC-08-1004 and IACUC 2010-13-2).
Allergen-induced airway inflammation
WT and NOX2 KO mice at the ages of 8 to 10 wk were divided into two groups and intraperitoneally injected with OVA (20 mg/mouse OVA in 2 mg aluminum hydroxide) on days 0 and 14. Mice were subsequently administered with either PBS or OVA (1% (w/v) in PBS) by intranasal route on days 28, 29, and 30. At day 32, mice were sacrificed to characterize the development of airway inflammation as reported (16).
Analysis of bronchoalveolar lavage fluid (BALF)
Mice were euthanized 48 h after the last OVA challenge. A tracheostomy was performed and the large airways were washed with 1 ml Hank's Buffered Salt Solution (Invitrogen, Carlsbad, CA, USA). The BALF was centrifuged at 4℃ at 200 g for 20 min and gently resuspended in PBS. The supernatant fractions were collected and stored at -70℃. Total cells numbers of BALF were counted in a HEMAVET (Drew Scientific Inc., Oxford, CT, USA). In addition, differential BALF cell counts were determined by performing Giemsa-stained cytospin preparations. Cells were counted at least five times using hemocytometer after exclusion of dead cells via trypan blue staining.
Histological analysis
After removal of BAKF, lung tissues were fixed in 10% (w/v) neutral buffered formalin for 24 h and embedded in paraffin. Tissue blocks were section at 10µm thickness, and stained with hematoxylin (Sigma-Aldrich Inc., MHS-16) and eosin (Sigma-Aldrich Inc., HT-110-1-32). Lung tissue sections were separately incubated with either periodic acid-Schiff (PAS, IMEB Inc., San Marcos, CA, USA) or Masson's trichrome staining solution (Sigma-Aldrich Inc., HT15) for measuring mucus secretion and collagen deposition, respectively.
Enzyme-linked immunosorbent assay (ELISA)
The EIA/RIA plates (Corning Inc., Corning, NY, USA) were pre-coated with purified Ab against IL-17 or IFNγ diluted in PBS at 4℃ overnight. The plates were incubated with blocking solution (10% FBS), followed by washing and incubating with cell supernatants overnight. The plates were subsequently washed with PBS and incubated with biotin-labeled anti-IL-17 or anti-IFN-γ Ab for 1 h. The plates were then washed and incubated with alkaline phosphatase-conjugated streptavidin for 30 min. The color changes of the plates were measured at 405 nm by an ELISA plate reader (Molecular Devices, Palo Alto, CA, USA).
Reverse transcription and quantitative real-time PCR
Total RNA was prepared and subjected to reverse transcription by Superscript II reverse transcriptase (Invitrogen). Quantitative real time-PCR was performed by ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA) using specific primer sets: eotaxin-FWD, 5'-cagatgcaccctgaaagccata-3', eotaxin-REV, 5'-tgctttgtggcatcctggac-3'; IL-17-FWD, 5'-caggacgcgcaaacatga-3', IL-17-REV, 5'-gcaacagcatcagagacacagat-3'; β-actin-FWD, 5'-agagggaaatcgtgcgtgac-3', and β-actin-REV, 5'-caatagtgatgacctggccgt-3'. Relative gene expression level was calculated after normalization to the level of β-actin.
Statistical analysis
All experiments were performed in triplicate or at least three times independently. The results were given as the mean±SEM. Statistical significance of the results was calculated by one-way analysis of variance and unpaired Student's t-test. p values of less than 0.05 are considered statistically significant.
RESULTS
Allergen injection augments red blood cell infiltration in NOX2-deficient lung
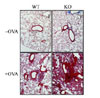
In order to clarify the susceptibility of NOX2 to airway inflammation, we introduced allergen-induced airway inflammation in wild type (WT) and NOX2 knockout mice (KO) by injecting OVA. First, we collected cells infiltrated into BALF and counted total cell numbers. While PBS-injected lungs reveal no significant infiltration of the cells, total infiltrated cells were increased by OVA injection but much higher in KO mice compared to WT mice (Fig. 1A). Particularly, red blood cells were highly increased in the lung of OVA-injected KO mice (Fig. 1B). Consistently, lung tissue staining revealed increased eosinophil infiltration in NOX2-deficient lung, however no critical difference between WT and KO in the absence of OVA challenge (Fig. 1C).
Immune cell infiltration and goblet cell hyperplasia are profound in NOX2 KO mice
Since OVA injection significantly increased immune cell infiltration including red blood cells in NOX2 KO mice, we further examined the immune cell types infiltrated into the lung. Numbers of neutrophils, macrophages, lymphocytes, and eosinophils were higher in KO mice than in WT mice, whereas the basophil number was comparable between two groups (Fig. 2A). Furthermore, histological analysis of lung verified the infiltrated immune cells and hyperproliferation of mucus-secreting goblet cells by challenge of OVA (Fig. 2B). However, Goblet cell hyperplasia stained by PAS was profound in the lung of NOX2 KO mice (Fig. 2B).
Airway remodeling induced by collagen deposition was prominent in NOX2-deficient mice
As collagen accumulation in the airway causes airway remodeling in chronic airway inflammation such as asthma, we next explored the collagen deposition without and with OVA challenge. Interestingly, NOX2 KO mice revealed more collagen deposition compared to that of WT lung even in the absence of OVA injection (Fig. 3), suggesting a tendency to airway inflammation in NOX2 KO mice. Moreover, OVA injection induced collagen deposition in WT mice, but drastically augmented in NOX2 KO mice. This result is in accordance with increased development of airway inflammation in NOX2 KO mice.
Inflammatory cytokines and eotaxin were enhanced in NOX2-deficient lung
Findings that NOX2 deficiency enhanced allergen-induced airway inflammation and collagen deposition in the airway prompted us to examine the cytokine production. T cells are primarily responsible for cytokine production in allergic airway inflammation. Thus, we analyzed the inflammatory T cell cytokines in the lung. Reverse transcription and real time-PCR analyses proved that IL-13 and IL-17 cytokines were significantly increased in NOX2-deficient lung (Fig. 4A and B). As the eosinophil recruitment was increased, the eosinophil-recruiting chemokine, eotaxin was elevated in deficiency of NOX2 (Fig. 4C).
Peripheral NOX2-null T cells augmented IL-17 and IFN-γ production upon TCR stimulation
Since IFN-γ and IL-17 expression were enhanced in NOX-deficient lung, we further analyzed cytokine production by T cells in OVA-challenged WT and KO mice. We isolated CD4+ T cells which are major cytokine producing cells and stimulated the cells with the plate-bound anti-CD3 Ab for 24 h in vitro. Supernatant of the stimulated cells was assayed for measuring T cell cytokines such as IL-17, IL-4, IL-5, IL-13, and IFN-γ. Interestingly, IL-17 and IFN-γ produced by CD4+ T cells were greater in KO CD4+ T cells than in WT cells (Fig. 5A and B). However, allergic cytokines including IL-4, IL-5, and IL-13 were not detected because of their limited production. These results indicate that NOX2-deficient CD4+ T cells are likely involved in inflammatory cytokine production, causing induced airway inflammation.
DISCUSSION
Our results demonstrate that NOX2 KO mice were more susceptible to development of airway inflammation upon antigen challenge and sensitization compared to WT mice. Immune cell infiltration, goblet cell hyperplasia, and collagen accumulated airway remodeling were prominently found in the lung of OVA-injected NOX-deficient mice. Consistently, inflammatory cytokines were significantly elevated in the lung and in the peripheral T cells of KO mice.
Allergen-induced inflammation recruits various kinds of immune cells including neutrophils, macrophages, monocytes, and lymphocytes in the inflammatory lung. Interestingly, NOX2-deficient mice significantly increased the infiltration of red blood cells into the lung, accompanying hemolytic anemia. Imbalance between reduced glutathione and oxidized glutathione through NADPH modulation causes weakening of the cell walls of red blood cells. The deficiency of NOX2 affects erythrocyte cell survival, resulting in hemolytic anemia. Recently, erythrocytes are reported to be critical for the regulation of hypoxia-induced lung inflammation (17). Hemoglobin autoxidation derives red blood cells to generate superoxide that readily changed to hydrogen peroxide, whereas hypoxia-induced responses were impaired in the absence of ROS generation in red blood cells. It remains to be clarified whether NOX2 is essential for the control of reduced glutathione levels and hemoglobin autoxidation.
We have recently reported that NOX2 deficiency spontaneously developed arthritis upon aging and with female preference (10). NOX2-deficient CD4+ T cells failed to produce Treg cells, whereas all effector Th1, Th2, and Th17 cells were all increased in NOX2 KO mice. As CD4+ T cells differentiate into regulatory T cells to control the immune reactivity, ROS generation seems to be important for sustaining the FoxP3-positive Treg cell development. Simultaneously, NOX2-deficient CD4+ T cells preferentially differentiated to IL-17-producing effector cells in vitro. We have also found that IL-17 production was prominently increased in allergen-induced inflammatory lung in NOX2 KO mice, but Th2 cytokines were expressed but comparably in WT and KO mice by injection with OVA. These results support that NOX2 functions in a T cell-intrinsic manner although NOX2 is known as an important regulator for phagocytic cells. Interestingly, NOX2 KO mice which developed severe arthritis developed more severe airway inflammation in response to OVA stimulation.
Our results confirm that NOX2 deficiency significantly increased inflammatory cytokine production in T cells and enhanced the susceptibility to development of allergen-induced airway inflammation thereby resulted in severe lung inflammation. It is likely that imbalance of ROS level in NOX2 deficiency may cause inflammation in multiple tissues in a T cell-autonomous manner.




 XML Download
XML Download