

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mycobacterium tuberculosis (Mtb) or mycobacterial Ags initiate proinflammatory responses in mononuclear phagocytes by interaction with a variety of signaling molecules/machineries. The relationships between specific mycobacterial components and signaling pathways are crucial for the induction of appropriate host responses, such as mounting a protective immune response, during mycobacterial infection (1). One of the Mtb-Ags, heparin-binding hemagglutinin (HBHA), is important for adherence to epithelial cells (2). This protein can induce mycobacterial aggregation, hemagglutination, and binding to heparin sulfate proteoglycans through post-translational modifications in the C-terminal lysine-rich domain of the protein (3,4). In addition, HBHA protein reacts with sera from tuberculosis (TB) patients, but not from healthy individuals, suggesting potent immunogenicity during mycobacterial infections (4). Moreover, HBHA has been widely studied for its potential to elicit effective host immune responses against TB (5). However, little is known regarding the molecular mechanisms by which HBHA modulates host innate immune responses.
Upon mycobacterial infection, the engagement of mycobacteria with innate receptors in host cells leads to the activation of signaling pathways that then results in the induction of inflammatory cytokines and antimicrobial effectors (6). Importantly, the mycobacterial cell wall contains a number of proinflammatory TLR2 ligands, including lipoproteins and lipoarabinomannan, and induces activation of the MAPKs and NF-κB pathways (1,7). Subsequent innate responses likely serve to decrease the inoculum size during the initial infection and drive an adaptive immune response (6). The MAPK pathways play an important role in enhancing antimycobacterial activity and production of immune effector molecules, including TNF-α (1). The proinflammatory cytokine TNF-α plays a crucial role in defense against mycobacterial infection including phagocytosis, intracellular killing, stimulating T cell activation, and granuloma formation (8,9). Our previous studies have reported that the proinflammatory cytokines TNF-α, IL-1β, and IL-6 are up-regulated in monocytes and macrophages isolated from the early stages of active pulmonary TB patients (10,11). In addition, it was shown that activation of ERK1/2 and p38 MAPK, proinflammatory cytokine secretion, and apoptotic activities were greater in monocytes or neutrophils from TB patients compared with healthy control subjects (11-13).
Although previous studies of TB and mycobacterial Ags have contributed to marked advances in our knowledge of the host protective immune responses, a number of critical questions are not well understood. Here, we present the intracellular signaling pathways activated by HBHA stimulation in murine bone marrow-derived macrophages (BMDMs). First, we determined whether HBHA Ag induced TNF-α and IL-6 production in BMDMs. We then examined the roles of PI3-K and MAPKs:p38, JNK, and ERK1/2 in BMDMs. We also found that the PI3-K and MAPK pathways contribute to an induction of HBHA-induced TNF-α and IL-6 production in BMDMs. To the best of our knowledge, this is first study to investigate the molecular mechanisms underlying the regulation of innate immune responses induced by HBHA.
MATERIALS AND METHODS
Mice and cell culture
Wild-type C57BL/6 mice were purchased from KOATECH (Pyungtek, Korea). All animal procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee, Chungnam National University. BMDMs were differentiated for 5~7 d in media containing M-CSF (R&D systems, Minneapolis, MN, USA), as described previously (1). The culture media contain DMEM (Gibco-BRL, Gaithersburg, NY, USA) with 10% heat-inactivated FBS (Gibco-BRL), 1 mM sodium pyruvate, 50 U/ml penicillin, 50µg/ml streptomycin and 5×105 M β-mercaptoethanol, sodium pyruvate, non-essential amino acids, penicillin G (100 IU/ml), and streptomycin (100µg/ml). The mouse macrophage cell line RAW264.7 (American Type Culture Collection; ATCC TIB-71) was maintained in complete medium (DMEM with 10% FBS) with sodium pyruvate, non-essential amino acids, penicillin G (100 IU/ml), and streptomycin (100µg/ml).
Reagents, DNA, and Abs
All reagents and chemicals were purchased from Sigma-Aldrich except those noted below, which were obtained from the indicated suppliers:BAY 11-7082, LY294002, Wortmannin, U0126, SB203580, and SP600125 were purchased from Calbiochem (San Diego, CA, USA). Specific Abs against total Akt, phospho-Akt (Ser473), phospho-IκB-α (Ser32/36), phospho-ERK1/2 (Thr202/Tyr204), phospho-p38 (Thr180/Tyr182), and phospho-JNK (Thr183/Tyr185) were purchased from Cell signaling Technology (Cell signaling, Beverly, MA, USA). Anti-β-actin (I-19), anti-IκB-α (C-21), and anti-phospho-NF-κB p65 (C-20) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). DMSO (Sigma, St. Louis, MO, USA) was added to cultures at 0.1% (v/v) as a solvent control. The dominant negative (DN)-p38, DN-MEK1 and DN PI3-K p110α constructs were a kind gift from Dr. Eui-Ju Choi (Korea University, Seoul, Korea). Cells were transfected using LipofectAMINE as indicated by the manufacturer (Invitrogen, Carlsbad, CA, USA).
Purification of recombinant HBHA
Methylated, recombinant HBHA was expressed in M. smegmatis (rMS-HBHA), as previously described with slight modifications (14,15). Purified recombinant HBHA was dialyzed against PBS (pH 7.2), applied to a column with immobilized polymyxin B (Detoxi-Gel Endotoxin Removing Gel; Pierce, Rockford, IL, USA) to reduce the level of endotoxin, filter-sterilized, and then frozen at -20℃. Protein concentrations were estimated using the bicinchoninic acid protein assay kit (Pierce), with BSA as the standard. The endotoxin content in HBHA was 0.017 ng/µg protein, as determined by the limulus-amebocyte-lysate test (QCL1000; BioWhittaker, Walkersville, MD, USA).
ELISA assay and Western blot
BMDMs were treated for the times indicated and processed for analysis by a sandwich ELISA and Western blot, as previously described (1). For the sandwich ELISA, cytokine levels in cell culture supernatants were determined using Duoset Ab pairs (BD PharMingen, San Diego, CA, USA) for the detection of TNF-α and IL-6, as described (1). All assays were performed as recommended by the manufacturers. For Western blot analysis, primary Abs were used at a 1:1,000 dilution. The blots were then detected by the ECL detection system according to the recommended procedure (Amersham-Pharmacia, Freiburg, Germany).
RNA isolation and RT-PCR assay
For semi-quantitative RT-PCR analysis, total RNA was extracted from BMDMs (1×106) using TRIzol (Invitrogen), as previously described (11). For PCR amplifications of specific cDNAs derived from TNF-α, IL-6, and β-actin mRNAs, three sets of sense and antisense oligonucleotides were used. Primer sequences were as follows:mTNF-α (forward:5'-CGGACTCCGCAAAGTCTAAG-3', reverse:5'-ACGGCATGGATCTCAAAGAC-3'), mIL-6 (forward:5'-GGAAATTGGGGTAGGAAGGA-3', reverse:5'-CCGGAGAGGAGACTTCACAG-3'), and β-actin (forward:5'-TCATGAAGTGTGACGTTGACATCCGT-3', reverse:5'-CCTAGAAGCATTTGCGGTGCACGATG-3'). PCR amplification was performed with an initial denaturing step at 95℃ for 5 min, followed by 30 cycles of denaturation (95℃, 1 min), annealing (58℃, 1 min), and extension (72℃, 1 min), then a further extension at 72℃ for 10 min. The PCR products were electrophoresed in 1.5% agarose gels in the presence of ethidium bromide, visualized by ultraviolet fluorescence, and recorded by a digital camera connected to a computer (SYNGENE, Gene Genius Imaging System).
Immunofluorescence microscopy for NF-κB translocation
For NF-κB translocation analysis, BMDMs were treated with HBHA for 1 h and then fixed with 4% (w/v) paraformaldehyde in PBS for 10 min at room temperature (RT). Cells were then permeabilized with 0.25% (v/v) Triton X-100 in PBS for 15 min at RT and blocked for 1 h in PBS containing 3% BSA. The cells were immunostained with rabbit anti-mouse NF-κB p65 (1:200 dilution) for 2 h at RT. After washing with PBS, cells were incubated with 1:200 dilutions of anti-rabbit immunoglobulin-conjugated AlexaFluor 488 (Molecular Probes, Eugene, OR, USA) for 1 h at RT. Nucleus were visualized by incubation with DAPI (1µg/ml, Sigma). The nuclear translocation of NF-κB p65 was visualized using a Olympus BX51 fluorescence microscope (DP ver. 1.2.1.108, Olympus, Japan).
RESULTS
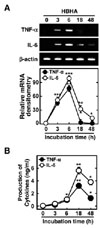
HBHA induces the expression of TNF-α and IL-6 mRNA and protein in BMDMs
Several mycobacterial Ags, including methylated HBHA, play an essential role in the induction of immunogenicity during tuberculous pleurisy infection (10-12). We first examined whether purified mycobacterial HBHA induced the expression of TNF-α and IL-6 mRNA in BMDMs. Semiquantitative RT-PCR analysis showed that HBHA significantly induced the expression of TNF-α and IL-6 mRNA after 3 h of stimulation in BMDMs. Peak expression of TNF-α and IL-6 mRNA occurred 6 h after stimulation with HBHA (Fig. 1A). In addition, secretion of TNF-α and IL-6 protein was assayed in BMDMs after HBHA stimulation. Treatment of BMDMs with HBHA significantly induced production of TNF-α and IL-6 after 6 h with a peak at 18 h (Fig. 1B). These data demonstrate that HBHA strongly induces proinflammatory cytokine expression by BMDMs.
HBHA robustly activates the NF-κB and MAPK signaling pathway in macrophages
NF-κB is a central regulator of inflammatory responses, and activation of NF-κB is required for the transcriptional induction of many proinflammatory mediators involved in innate immunity, including cellular adhesion molecules, cytokines, and growth factors (16). After stimulation with HBHA for the times indicated, expression of IκB-α was dramatically attenuated after 15~30 min, whereas IKKα/β phosphorylation was strongly induced after 15~30 min (Figs. 2A and B). The MAPK pathways are crucial for macrophage signaling during mycobacterial infection (17,18). We next examined whether HBHA induced MAPK activation in BMDMs. HBHA stimulation induced MAPK (ERK 1/2, p38, and JNK) activation within 15 min of stimulation, with a peak at 15~30 min (Figs. 2A and C). These data indicate that HBHA robustly activates NF-κB and MAPK signaling pathways in macrophages.
NF-κB activation is required for HBHA-induced TNF-α and IL-6 production
We next investigated the role of NF-κB in HBHA-induced proinflammatory cytokine production by BMDMs. We first confirmed translocation of NF-κB p65 into the nucleus after stimulation with HBHA (Fig. 3A). Pretreatment of BMDMs with an NF-κB inhibitor (BAY 11-7082, an inhibitor of IκB phosphorylation) significantly attenuated production of TNF-α and IL-6 in a dose-dependent manner (Fig. 3B). Furthermore, pre-treatment of BMDMs with other NF-κB inhibitors (IKK-2 inhibitor and NF-κB activation inhibitor) significantly reduced TNF-α and IL-6 mRNA expression (data not shown). These results indicate a critical role for NF-κB in the modulation of HBHA-induced inflammatory responses.
HBHA-induced TNF-α and IL-6 expression is dependent on the PI3-K/Akt pathways
In previous studies, we found a rapid phosphorylation of the PI3-K/Akt pathway in human monocytes after stimulation with the 30 kDa or purified protein derivative (PPD) of Mtb (19,20). However, the nature of PI3-K/Atk activation after HBHA stimulation remains unknown. We also investigated whether HBHA stimulation induces activation of the Akt pathway in BMDMs. As shown in Fig. 4A, HBHA induced phosphorylation of Akt within 5 min of stimulation and marked activation of Akt occurred within 2~8 h of HBHA stimulation.
To examine the role of the PI3-K/Akt pathway in HBHA-induced TNF-α and IL-6 production, BMDMs were pre-treated with LY294002 or Wortmannin, which are pharmacologic inhibitors of PI3-K, prior to HBHA stimulation. TNF-α and IL-6 mRNA and protein levels in the cells and supernatants, respectively, were then examined. As shown in Figs. 4B and C, pre-treatment with LY294002 or Wortmannin significantly inhibited TNF-α and IL-6 mRNA expression (Fig. 4B) and protein production (Fig. 4C) in HBHA-treated BMDMs. Furthermore, these data were confirmed using PI3-K p110α-DN. RAW264.7 cells were transfected with the PI3-K p110α-DN construct or mock control before stimulation by HBHA and then subjected to ELISA. A similar significant inhibition of TNF-α and IL-6 production was observed in RAW264.7 cells transfected with the PI3K p110α-DN mutant construct compared with the mock control vector (Fig. 4D). These data suggest that the PI3-K/Akt pathway is essential for regulation of HBHA-induced TNF-α and IL-6 production by macrophages.
HBHA-induced TNF-α and IL-6 expression is dependent on the MAPK pathway
We next investigated the possible involvement of the ERK 1/2, p38 MAPK, or JNK pathways in HBHA-induced proinflammatory responses. Cells were stimulated with HBHA and cultured for 6 or 18 h to assess mRNA expression and TNF-α and IL-6 production. As shown in Figs. 5A and B, HBHA-induced TNF-α and IL-6 expression and protein production were significantly inhibited in BMDMs pretreated with specific MAPK inhibitors [inhibitors of MEK (U0126, PD98059), p38 MAPK (SB203580), and JNK (SP600125)].
Additionally, RAW264.7 cells were transfected with p38 MAPK-DN and MEK1-DN mutant constructs or a mock control vector prior to stimulation by HBHA. We observed a significant inhibition of TNF-α and IL-6 production in RAW264.7 cells transfected with the p38 MAPK-DN or MEK1-DN mutant construct compared with the mock control vector (Fig. 5C). These results indicate that HBHA-induced TNF-α and IL-6 production is regulated by the MAPKs (ERK 1/2, p38 MAPK, and JNK) pathway in macrophages.
PI3-K inhibition attenuates HBHA-induced ERK1/2 and p38, but not JNK, activity in BMDMs
Mycobacteria and mycobacterial components are potent activators of the PI3-K/Akt and MAPK pathways in macrophages (17,18,21-23). Previously, it was shown that the PI3-K and Akt pathways modulate the activation of ERK1/2 in response to Mtb or mycobacterial Triton X-100-solubilized protein Ag (24). We further determined whether HBHA-dependent PI3-K activation regulates the downstream MAPK pathways. To investigate the role of PI3-K in HBHA-induced MAPK activation, BMDMs were pretreated with Wortmannin or LY294002, stimulated with HBHA, and then assessed by Western blot analysis for ERK1/2, p38 MAPK, and JNK phosphorylation. As shown in Fig. 6, inhibition of PI3-K significantly attenuated HBHA-induced phosphorylation of Akt, as well as that of ERK1/2 and p38, in BMDMs. However, the activation of JNK was not modulated by inhibition of the PI3-K pathway. These results show that PI3-K/Akt activity is required for the HBHA-induced activation of ERK 1/2 and p38 MAPK, but not JNK.
DISCUSSION
In the present study, we demonstrate the molecular mechanisms underlying the regulation of the proinflammatory cytokines TNF-α and IL-6 by the PI3-K/Akt, p38, and ERK 1/2 MAPK pathways in response to the mycobacterial HBHA in murine macrophages. Mycobacteria and their components are potent activators of macrophages, which are able to produce proinflammatory mediators that are essential for innate defense and induction of the acquired immune response against mycobacteria (17,21,25,26). The mycobacterial HBHA, especially the methylated form, is known to be essential for effective protective immunity, which is comparable to that induced by vaccination with Bacillus Calmette-Guérin (27). Furthermore, previous studies reported that HBHA is significantly more sensitive than early secreted antigenic target-6 and more specific than PPD for the detection of latent TB infection (28). These observations strongly suggest that mycobacterial HBHA is involved in the pathogenesis of TB and might be useful in diagnosis. However, the role of HBHA in the induction of innate and inflammatory responses during mycobacterial infection has not been characterized.
We found that levels of the proinflammatory cytokines TNF-α and IL-6 were increased in BMDMs stimulated with HBHA. Vaccination with HBHA induces IFN-γ production by both CD4+ and CD8+ T cells, and CD8+ cytotoxic T cell responses (29). To the best of our knowledge, this is the first report that HBHA actively induces proinflammatory immune responses in macrophages. TNF-α, a critical proinflammatory cytokine, plays an essential role in the host protective immune responses against mycobacterial infection (30). We previously reported that mycobacterial Ags up-regulated the production of TNF-α, IL-1, and IL-6 in monocytes/macrophages isolated from early active TB patients, when compared with those from healthy controls (10,11). In addition, activation of ERK1/2 and p38 MAPK, proinflammatory cytokine secretion, and apoptotic activities were greater in monocytes or neutrophils from TB patients, compared with healthy control subjects (11-13). Combined with our findings, these reports suggest that HBHA contributes to the immunopathology of TB through active induction of proinflammatory cytokine release.
NF-κB is a central mediator of the inducible transcription of various proinflammatory genes in innate immune responses (31). Previous studies have shown that mycobacterial infection, or mycobacterial products, act through TLRs to trigger the MAPK pathways, leading to activation of transcription factors, including NF-κB (17,24,32). We found HBHA-induced increases in IKKα/β phosphorylation, as well as translocation of p65 from the cytosol to the nucleus. Importantly, inhibition of NF-κB significantly reduced HBHA-induced proinflammatory cytokine secretion. These data are consistent with the observed strong induction of proinflammatory cytokine expression by heat shock protein (33) and a 19-kDa lipoprotein (34) from Mtb. Taken together, our data suggest that NF-κB activation plays an essential role in HBHA stimulation of proinflammatory cytokine secretion by BMDMs.
Mycobacteria induce the PI3-K (35,36) and MAPK intracellular signaling cascades (1,21). In mycobacterial infections, the PI3-K pathway plays a role in human monocyte antimycobacterial activity (11,37). The MAPK signaling pathways are activated upon mycobacterial infection, and have been implicated in pathogenesis (18). The mycobacterial Ags 38 kDa and MTB12 can induce activation of ERK1/2 and p38 MAPK, and subsequent cytokine secretion in monocytes from active pulmonary TB patients (11,38). Although HBHA is widely studied for its potential to trigger effective host immune responses against TB (5), little is known regarding the signaling mechanisms underlying proinflammatory cytokine secretion by macrophages. Here, we suggest that mycobacterial HBHA can induce intracellular signaling pathways, such as PI3-K and MAPK, that are required for production of TNF-α and IL-6 in BMDMs.
Previously, we reported that macrophage recognition of mycobacteria and their Ags via innate receptors resulted in the activation of the MAPK and PI3-K/Akt pathways in monocytes/macrophages (11,21,39). The PI3-K and MAPK pathways contribute to proinflammatory cytokine secretion in response to the PPD and 30 kDa Ag of Mtb in human monocytes (19,20). Additionally, the PI3-K/Akt and ERK1/2 pathways are important for TNF-α expression in human monocyte-derived macrophages after treatment with Mtb H37Rv and Triton X-100-solubilized protein purified from Mtb (24). Interestingly, HBHA elicited a biphasic response, with an early peak of phospho-Akt observed after 5 min of stimulation, followed by a drop at 30~60 min and then a steady increase up to 8 h. These data are partly consistent with previous studies in which PI3-K/Akt showed a similar biphasic activation, i.e., by Francisella infection in murine macrophages (40) and LPS or α-lipoic acid in human monocytic THP-1 cells (41,42). Currently, the mechanisms by which phosphorylation of Akt at Ser473 are modulated during the early and late phases after stimulation of HBHA are not known. Future studies will likely elucidate the molecular mechanisms by which Akt phosphorylation is modulated and the exact roles of Akt phosphorylation during the early and late phases after HBHA stimulation.
Our results also demonstrate that PI3-K is a necessary upstream activator of the p38 and ERK1/2 MAPK pathways in BMDMs after stimulation with HBHA. Crosstalk between PI3-K/Akt and MEK-ERK pathways in different cell types has been demonstrated (43). Previously, it was reported that the PI3-K pathway is an upstream signal for ERK1/2 MAPK activation (24). However, this study did not determine how important HBHA is in proinflammatory innate responses or in the pathogenesis of TB. However, our data suggest a potential role for HBHA in the proinflammatory response to mycobacterial infection, since HBHA is a strong stimulator of proinflammatory cytokine secretion and an NF-κB activator. Recently, Jung et al. demonstrated that HBHA up-regulates the proinflammatory cytokines IL-6, IL-12, IL-1β, TNF-α, and CCR7 in dendritic cells (44). In the same study, HBHA-treated dendritic cells activated naïve T cells and polarized them to secrete IFN-γ (44). Other studies have shown that HBHA-induced IFN-γ production in alveolar and pleural lymphocytes is higher in pulmonary or pleural TB patients than in non-TB controls (45). Moreover, HBHA is strongly recognized by sera from pulmonary TB patients when compared with healthy controls (46). Taken together, our data strongly suggest that HBHA plays a potentially pleiotropic function in protective immunity (through IFN-γ-dependent protective immunity) and inflammatory responses (through inflammatory cytokine production) in TB.
Collectively, the data presented in this study provide a novel insight into molecular signaling by HBHA through activation of the NF-κB and PI3-K/Akt-p38-ERK1/2 MAPK pathways, which are responsible for the induction of pro-inflammatory responses during TB infection. In addition, our data reveal the key immunological processes induced by important human pathogens, including mycobacteria, and this information may assist in the rational design of more effective vaccines and adjuvants.





 XML Download
XML Download