

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Liver disease is one of major causes of morbidity and mortality worldwide and alcohol is one of the most prominent factors in liver disease (1,2). Alcoholic liver disease (ALD) encompasses a broad spectrum of diseases ranging from steatosis (fatty liver), steatohepatitis, fibrosis, cirrhosis to hepatocellular carcinoma (3,4). Of the ALD, steatosis is considered as mild condition, whereas steatohepatitis is pathogenic condition, which has a potential of progression to liver fibrosis, cirrhosis or hepatocellular carcinoma (5). Generally, the liver is composed of parenchymal cells (e.g. hepatocyte) and non-parenchymal cells such as sinusoidal endothelial cells, Kupffer cells, hepatic stellate cells (HSCs), dendritic cells and other lymphocytes. Interestingly, liver comprises enrichment of innate immune cells such as resident macrophages (Kupffer cells), natural killer (NK), NKT and γδ T cells. (6-8). For example, mouse liver lymphocytes contain 10% NK cells, whereas rat and human liver lymphocytes contain about 30% to 50% NK cells. NKT cells constitute up to 30% and 10% of the intrahepatic lymphocyte population in mice and human respectively (6,7,9). Interestingly, ALD has been considered as complex consequences of interaction among these cells, especially between hepatocytes and hepatic stellate cells or innate immune cells (3,4). Previous our studies demonstrated that alcoholic liver steatosis was induced by HSC-mediated endocannabinoid and its hepatic CB1 receptor (10) and alcoholic liver fibrosis was accelerated due to abrogation of antifibrotic effects of NK cells/interferon-γ (IFN-γ) against HSCs (11). Other studies suggest that Kupffer cells are mainly involved in alcohol-mediated inflammation via lipopolysaccharide (LPS)/toll like receptor 4 (TLR4) signaling-dependant mechanisms (4,12). Based on these evidences, researchers recognized the importance of innate immune response in ALD. Therefore, in this review, we will focus and discuss about diverse involvements of HSCs and innate immunity (Kupffer cells/macrophages and NK cells) in ALD such as alcoholic steatosis, steatohepatitis and liver fibrosis.
ALCOHOLIC LIVER STEATOSIS (FATTY LIVER)
Alcoholic liver steatosis has been considered as a weak condition for a long time. However, increasing evidence suggests that it seems to be a potentially pathologic state and it will progress more severe state in the presence of other co-factors such as sustained consumption of alcohol, hepatic virus infection, diabetes or drugs (13,14). Fat accumulation in hepatocytes is the result of imbalanced fat metabolism such as decrease mitochondrial lipid oxidation but enhanced synthesis of triglycerides. Several underlying mechanism of this have been suggested that it might be related with increased NADH/NAD+ ratio (15,16), increased sterol regulatory element-binding protein-1 (SREBP-1) activity (17,18), decreased peroxisome proliferator-activated receptor-α (PPAR-α) activity (19,20) and decreased AMP-activated protein kinase (AMPK) activity (10,18).
In addition to pathological mechanisms for this, there are several interesting evidences that innate immune cells of liver are related in alcoholic liver steatosis (3,4,12,21,22). Kupffer cells are one of the main innate immune cells involved in steatotic liver. First, alcohol increased gut permeabilization, which allows more uptake of endotoxin/LPS in portal circulation (21) and then Kupffer cells are activated in response to LPS via TLR4 signaling cascade, leading to produce several kinds of pro-inflammatory cytokine such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1, IL-6 and reactive oxygen species (ROS) (3,4,22). Of these cytokines, contribution of TNF-α in developing alcoholic liver steatosis has been less characterized compared with TNF-α-mediated hepatic inflammation. However, increased expression of TNF-α has been observed in alcoholic liver steatosis of mouse (23,24) and absence of its receptor (TNF-α R1) activity inhibits the development of alcoholic liver steatosis (25,26). In addition, it has been reported that TNF-α has a potential to increase mRNA expression of SREBP-1c, a potent transcription factor of fat synthesis, in the liver of mice and to stimulate the maturation of SREBP-1 in human hepatocytes, respectively (27,28). Furthermore, recent report showed that alcohol-mediated infiltration of macrophages decreased amounts of adiponectin (known as anti-steatosis peptide hormone) production of adipocytes, leading to alcoholic liver steatosis (29). Therefore, Kupffer cells/macrophages may contribute to the development of alcoholic liver steatosis by upregulating the SREBP1 activity in hepatocytes and downregulating the production of adiponectin in adipocytes. In contrast, IL-6 produced by Kupffer cells may play a crucial role in protecting against alcoholic liver steatosis via activation of signal transducer and activator of transcription 3 (STAT3), consequently inhibiting of SREBP1 gene expression in hepatocytes (30-32).
More interestingly, a recent our study reported that alcoholic liver steatosis was mediated mainly through activated HSCs-derived endocannabinoid and its receptor (CB1R) of hepatocytes (10). This study suggested that chronic alcohol drinking activates HSCs to produce 2-arachidonoylglycerol (2-AG), one of endocannabinoid, which then increased the expression of SREPB1c and fatty acid synthase (FAS) but decreased AMPK activation, consequently leading to accumulation of fat in hepatocyte. Another studies reported that activated HSC can be killed by liver NK cells through IFN-γ and tumor necrosis factor-related apoptosis inducing ligand (TRAIL)-dependent manners (11,33-35). However, chronic alcohol consumption inhibited NK cytotoxicity against activated HSC, leading to prolonged survival of activated HSC (11).
ALCOHOLIC STEATOHEPATITIS (ASH)
Alcoholic steatohepatitis means united condition with fat accumulation and inflammation in the liver and more progressed pathologic state compared with alcoholic liver steatosis. In response to alcohol intake, innate immune cells initiate and maintain hepatic inflammation via pattern recognition receptors, especially TLRs (36-38).
Among activation of innate immune cells in liver, Kupffer cells have been identified as one of the significant elements in the pathogenesis of alcoholic steatohepatitis (22). Considering their specific location at the interface between the portal and the systemic circulation, Kupffer cells play a key role in orchestrating the immune response against endotoxin/LPS. LPS, a component of gram-negative bacteria wall, has been considered to be a key molecule of activation in Kupffer cells, in which signals are transmitted through TLR4 (3,38-40). TLR4 is a major component of the LPS recognition receptor complex, which also involves the co-receptors CD14 and MD-2, and LPS binding protein (LBP) (41,42). LBP is a soluble shuttle protein that directly binds LPS and facilitates the association between LPS and CD14 (43). Studies in knockout mouse models have shown that chronic alcohol feeding in mice deficient of CD14, TLR4 and LPS-binding protein (LBP) results in alleviation of alcohol-induced liver injury indicating an important role for the TLR4 pathway (40,44,45). In addition, the importance of gut-derived endotoxin/LPS in ALD was suggested by experiments where treating the animals either with antibiotics or with lactobacilli to remove or reduce the gut microflora provided protection from the features of ALD (46). TLR4 initiates two major pathways. After binding LPS with TLR4, recruited TIR domain-containing adaptor protein (TIRAP) and myeloid differentiation factor 88 (MyD88) lead to early-phase activation of nuclear factor-κB (NF-κB), producing pro-inflammatory cytokines including TNF-α, IL-6 and monocyte chemotatic protein-1 (MCP-1). Meanwhile TIR-domain containing adaptor inducing IFN-β (TRIF) and TRIF-related adaptor molecule (TRAM) activate interferon regulatory factor 3 (IRF3) leading to the production of type I IFN and late activation of NF-κB (38,47). However, there is a recent report that alcohol-mediated liver injury and inflammation are mainly induced by TLR4-dependent but MyD88-independent manners (48). Among pro-inflammatory cytokines, particularly TNF-α mainly contributes to the development of ALD and its level is increased in patients with ASH (22) and in the liver of alcohol-fed animals (23,24). In addition, Kupffer cells secrete other important cytokines, including IL-8, IL-12, and interferons, that contribute to the intrahepatic recruitment and activation of granulocytes that are characteristically found in severe ALD, influence immune system polarization (49). Interestingly, TLR4 is expressed not only on innate immune cells such as Kupffer cells and recruited macrophages, but also on hepatocytes, sinusoidal endothelial cells and stellate cells in the liver (39). The role of TLR4 in these cells has to be investigated.
In addition to LPS, oxidative stress-mediated cellular responses also play an important role in innate immune cell activation. Kupffer cells are also a major source of ROS in response to chronic alcohol exposure (50,51). One important ROS is the superoxide ion, which in activated phagocytes is mainly generated by the enzyme complex NADPH oxidase. Underscoring the important role of ROS in mediating ethanol damage, treatment with antioxidants and deletion of the p47phox subunit of NADPH oxidase in ethanol-fed animals could reduce oxidative stress, activation of NF-κB, and TNF-α release in Kupffer cells, thus, preventing liver injury (50,52). In addition, NADPH oxidase induces TLR2 and TLR4 expression in human monocytic cells (53). Furthermore, evidence shows that direct interaction of NADPH oxidase isozyme 4 with TLR4 is involved in LPS-mediated ROS generation and NF-κB activation in neutrophils (54).
ALCOHOLIC LIVER FIBROSIS
HSCs are known as a major cell type inducing liver fibrosis (55). Generally, HSCs are located in the Disse's space as a quiescent status in normal healthy liver, but by liver injuries they become activated and differentiated into myofibroblastic cells that are characterized by a loss of vitamin A (retinol) and enhanced extracellular matrix, especially collagen fibers, leading to liver fibrosis.
Interestingly, recent reports suggest that innate immune cells (Kupffer cells and NK/NKT cells) are closely involved in the liver fibrogenesis. First, Kupffer cells are generally considered as a positive regulator in liver fibrosis. Actually, increasing evidences suggest that chronic alcohol consumption leads to liver fibrosis via the activation of the profibrogenic effects of Kupffer cells due to enhanced transport of endotoxin/LPS in the gut, inducing production of pro-inflammatory cytokines such as TNF-α, IL-6 and transforming growth factor-β (TGF-β) (3,14,23,24). Among these cytokines, TGF-β is known as a key regulator of HSC activation and differentiation. Thus, it is believed that Kupffer cells positively regulate alcoholic liver fibrosis via TGF-β-mediated HSC activation and subsequently promote collagen synthesis of HSCs. In contrast, recent findings interestingly suggest the novel role of NK cells having anti-fibrotic effects via multiple mechanisms (33-35,56,57). First, NK cells directly kill early activated HSCs in NKG2D- and TRAIL-dependent manners but not quiescent HSCs (35,56). This is because early activated HSCs express NK cell-activating ligand retinoic acid early inducible gene 1 (RAE-1) via retinol metabolism and TRAIL receptors but express decreased MHC-I, NK cell-inhibitory ligand (33,57). Second, NK cells can suppress liver fibrosis via production of IFN-γ, which induces HSC cell cycle arrest and apoptosis in STAT1-dependant manner (34,58). Analogous to NK cells, NKT cells (invariant NKT cells) can also suppress activation of HSCs via direct killing and IFN-γ production (59). However, anti-fibrotic effects of NKT cells are beneficial only at early stage of liver fibrosis because of their quick depletion.
Although the functions of hepatic NK/NKT cells are known a little in ALD, it is generally accepted now that chronic alcohol consumption accelerates liver fibrosis due to the suppressed activity of NK cell in patients and mice and enhanced alcoholic liver injury as well (11,60-62). The activation status of NK cells has been reported to be decreased by alcohol, possibly because of increased IL-10 and TGF-β release by monocytes and activated HSCs (11,63) In case of NKT cells, they seem to contribute to alcoholic liver injury since activation of NKT cell accelerates whereas NKT deficiency delays alcoholic liver injury (64,65). Nevertheless reports of alcohol effects on NK/NKT cell functions are still controversial. Therefore, further investigation is needed in animal and human.
CONCLUSIONS
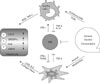
The innate immune cells of liver play important roles in dangerous stress such as alcoholic liver injuries and infection of microorganism. Recently a line of evidences has demonstrated that innate immune responses are closely connected with the development of ALD and we integrate these findings in Fig. 1. Especially, activation of Kupffer cells by alcohol appears to be required for the development of alcoholic steatohepatitis via LPS-TLR4 signaling pathways. However, endocannabinoid production (2-AG) of activated HSCs might be another major factor for the induction of alcoholic steatosis. Therefore, we should simultaneously consider both types of cells when developing therapeutics for alcoholic steatohepatitis. For example, although we successfully inhibit activation of Kupffer cells by drugs activated HSCs still induce accumulation of fat in the liver, leading to lipotoxicity and in turn to generative of oxidative stress and inflammation, subsequently restoring steatohepatitis. More interestingly, although the role of NKT in ALD is obscure, NK cells are responsible for anti-steatotic and anti-fibrotic functions via killing activated HSCs that may reduce production of 2-AG and collagen. However, functions of NK cells are abrogated or suppressed by chronic alcohol consumption and high level of TGF-β in the liver. Therefore, further understanding of the roles of innate immunity and HSCs help us to develop novel therapeutic targets to treat ALD.
 XML Download
XML Download