

 PDF
PDF ePub
ePub Citation
Citation Print
Print


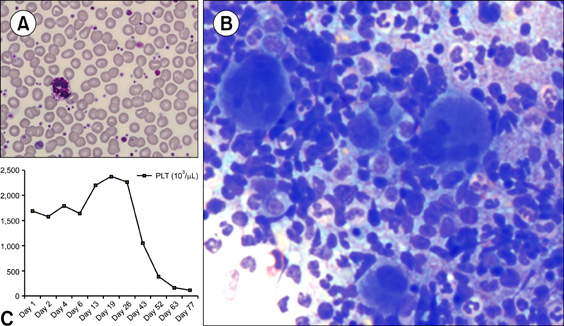
A 51-year-old man with no specific medical history was brought to our hospital with general weakness. Complete blood count results were as follows: Hb level, 12.4 g/dL; platelet count, 1,594×109/L; leukocyte count, 16.52×109/L; segmented neutrophils, 65%; lymphocytes, 26%; monocytes, 3%; eosinophils, 2%; and atypical lymphocytes, 4%. Peripheral blood smear showed marked increase in giant platelets and hypogranulated platelets (A). Bone marrow aspirate displayed normal erythropoiesis and increased granulopoiesis, and a notable finding was marked increase in the number of small megakaryocytes, namely, "dwarf megakarytocytes." However, a few megakaryocytes showed hyperplastic features (B). Essential thrombocythemia (ET) was preferentially considered over other myeloproliferative neoplasms (MPNs). However, both JAK2 V617F and MPL 515 mutations were not detected. Interestingly, the Philadelphia chromosome was detected in 19 out of 20 bone-marrow metaphase cells analyzed. Subsequently, BCR/ABL1 fluorescence in situ hybridization (FISH) analysis yielded a positive result. Reverse transcriptase- polymerase chain reaction (RT-PCR) analysis for BCR/ABL1 rearrangement showed the presence of the b3a2 fusion gene. Finally, the patient was diagnosed with chronic myeloid leukemia (CML) associated with extreme thrombocytosis (C). After one course of imatinib treatment, the patient's platelet count was reduced to normal levels. Although CML is easily predicted by approaches of morphologic basis, cases with extreme thrombocytosis would require molecular techniques such as chromosome, FISH and RT-PCR for a proper differential diagnosis including other disorders of MPN such as ET.
 XML Download
XML Download