

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Congenital factor VII (FVII) deficiency is an autosomal recessive disorder with an estimated prevalence of 1/500,000 individuals without ethnic or gender predilection [1, 2]. There are 31 known patients with congenital FVII deficiency in Korea [3]. This is a rare bleeding disorder, which ranges in clinical severity from life-threatening to asymptomatic [4-6]. The affected person may experience recurrent joint bleeding that may lead to chronic pain and joint impairment, with subsequent disability. Persons who are homozygous or double heterozygous for the trait usually have FVII levels that are less than 10% of normal, and heterozygotes typically have FVII levels of approximately 50% of the normal level. However, unlike with hemophilia A and B, there is only a weak correlation between FVII activity and bleeding risk [7, 8]. Treatment demands vary considerably for most FVII-deficient patients, ranging from none or rare on-demand treatment to a need for frequent factor administration for long- or short-term prophylaxis such as for major surgical interventions [9].
Surgery in patients with FVII deficiency has been reported to be endangered by intraoperative or postoperative bleeding, unless a replacement therapy is used [10]. In general, surgical bleeding is not an infrequent symptom in FVII deficiency, reported in approximately one-third cases [5]. It is generally believed that substitution therapy should be administered to patients undergoing surgery who have FVII activity of less than 10-15% and to patients with a history of recurrent bleeding [11].
Recombinant activated FVII (rFVIIa, NovoSeven®, Novo Nordisk, Bagsvaerd, Denmark) has been used in the last decade in patients with this coagulation disorder and causes effective hemostasis in most cases. Initially approved for use in hemophilia with inhibitors, rFVIIa has been approved to treat bleeding in patients with congenital FVII deficiency. However, the optimal treatment regimen, mode of administration, and target FVII activity remain underdetermined [12].
Antifibrinolytic agents are an additional pharmacological option in bleeding disorders. Tranexamic acid is an antifibrinolytic agent that competitively inhibits the activation of plasminogen to plasmin. It promotes clot stability and is useful as adjunctive therapy in hemophilia and some other bleeding disorders. It is valuable in controlling bleeding from the skin and mucosal surfaces (e.g., oral bleeding, epistaxis, and menorrhagia) [13].
Because FVII deficiency is a rare bleeding disorder, a limited number of patients are available for follow-up in most treatment centers. In this article, we review the results of surgical procedures in patients with congenital FVII deficiency at our center and describe the safety and effectiveness of surgery with rFVIIa or with an antifibrinolytic agent without rFVIIa.
MATERIALS AND METHODS
Five patients with congenital FVII deficiency, who had undergone 8 surgical procedures between January 2008 and June 2010, were included in this study. FVII deficiency may be easily suspected when a hemostatic screening reveals an isolated prolongation of the prothrombin time with a normal activated partial thromboplastin time. FVII activity was the confirmatory test for diagnosis. In order to evaluate subjects at a clear risk of bleeding, only patients with FVII activity of <10% of normal were included in this study (Table 1).
We retrospectively reviewed the electronic medical records of these 8 cases, including demographic characteristics, FVII activity, presence of previous symptomatic bleeding, previous history of rFVIIa injection, surgical procedure, rFVII therapy (replacement duration [number of days between the first and final day of rFVIIa therapy], total replacement dose, and mean daily dose [total dose/number of treatment days]). rFVIIa was administered in severe cases and when bleeding history was present, while asymptomatic patients undergoing minimally invasive surgical procedures such as dental extraction were treated with tranexamic acid-given orally at the usual dosage.
rFVIIa was administered intravenously with doses of 15.30 µg/kg every 4 hrs postoperatively during the 48 hrs following the first dose that was administered 1 hr preoperatively; thereafter, the dosing interval was adjusted according to the FVII activity and the patient's progress. FVII activity was measured at baseline, before rFVIIa administration, and 30 min after rFVIIa administration.
Tranexamic acid (Transamin®) was administered in the case of dental extraction, with a dose of 15.25 mg/kg 2 or 3 times a day.
RESULTS
Patient characteristics are summarized in Table 1. Five patients with congenital FVII deficiency underwent 8 surgical procedures. The median age of the patients was 19.5 years (range, 6.37 years). FVII activity ranged from 0.6% to 7%. Four were major surgeries and 4 were minor. All 4 major surgeries were orthopedic procedures. Of the 4 minor cases, 1 was a tonsillectomy and the remaining 3 were dental extractions. The median duration of hospitalization was 8.5 days (range, 0.15 days). The management and outcomes of the 8 surgical cases are summarized in Table 2.
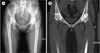
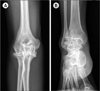
Of the 5 patients, 2 had a history of recurrent bleeding: a female patient (cases 2, 3, and 4) had menorrhagia and hemophilic arthropathy of the hips, ankles, and elbows (Fig. 1). She experienced frequent hemarthrosis and joint pain. The other patient (case 5) had hemarthrosis of the ankle and elbow and suffered from hemophilic arthropathy (Fig. 2). Arthroscopic synovectomy procedures for ankle and elbow were performed.
Of the 8 surgeries, rFVIIa was used in 6; however, it was not used for 2 dental extractions (cases 6 and 8). Among the 6 cases of rFVIIa therapy, the duration of days of rFVIIa therapy ranged from 1 to 15 days, and the total number of doses ranged from 1 to 42. The minimum and maximum total doses of rFVIIa were 30 and 960 µg/kg, respectively. After the initial bolus of rFVIIa infusion, the dosing interval was adjusted according to the FVII activity. FVII activity was measured at baseline and before and after the intravenous administration of rFVIIa. For example, in case 2, in which baseline FVII activity was 2%, FVII activity reached 310% at 30 min after an initial 24 µg/kg dose bolus infusion of rFVIIa and declined to less than 61% immediately before the next infusion (Fig. 1). Fig. 1 shows the FVII activity (%) and dosing regimen of rFVIIa in 3 cases (cases 1, 2, and 5). Dental extraction was performed with only antifibrinolytic agents without any replacement and no bleeding occurred (cases 6 and 8). In 1 case (case 7), the patient received both rFVIIa replacement therapy and antifibrinolytic therapy.
Severe bleeding was not reported during any of the 8 surgical interventions. No thrombotic episodes were observed during the surgery or the follow-up period.
DISCUSSION
Congenital FVII deficiency is associated with reduced levels of FVII activity; it is characterized by spontaneous bleeding episodes in severely affected patients and by bleeding after trauma or surgical intervention in milder cases. As described previously, the correlation between FVII activity and bleeding tendency is poor, although severe bleeding is most commonly associated between low FVII activity levels and the surgical risk of bleeding [14].
rFVIIa came on the market in 1996 and was a new specific treatment option for FVII-deficient patients, although the drug was developed primarily for use in hemophilia patients with an inhibitor. Replacement therapy is necessary for controlling hemorrhagic risk, but there are few guidelines for its management.
Fresh-frozen plasma (FFP) is still used in developing countries, but its use is associated with some important adverse outcomes such as blood volume overload and a relatively high risk of transmission of blood-borne viruses. Plasma-derived FVII concentrates are essentially prothrombin complex concentrates (PCCs) with a significantly higher content of FVII relative to the other vitamin K-dependent proteins. These are also effective for any therapeutic requirement, but these concentrates carry the risk of blood-borne virus transmission. According to 1 study, the comparison of potency (IU/mL) in these concentrates is 1 in FFP, 5.10 in PCCs, and >25,000 in rFVIIa [15].
A recent report on surgery in FVII-deficient patients covered using rFVIIa indicated that bolus injections were effective [16]. The target level of FVII activity for surgery in FVII-deficient patients has not yet been determined, although it has been a level of 10% as a threshold has been recommended, in combination with a full clinical history to determine the need for replacement therapy [11].
This study included patients with congenital FVII deficiency who underwent 8 surgical procedures and were administered either a bolus of rFVIIa or an antifibrinolytic agent. Our report comprises only interventions carried out with rFVIIa, a treatment that is becoming the most commonly used in FVII deficiency, or with an antifibrinolytic agent. Dental extractions (cases 6 and 8) were performed with only antifibrinolytic agents without any replacement. We found that excellent hemostasis was achieved for all procedures.
In general, the dose and dosing interval in FVII deficiency is initially 15.30 µg/kg every 4 hrs, and we also administered the drug in accordance with this policy. The response to factor administration was different for each patient. One patient's FVII trough level immediately before the next dose was 52%, which was under the normal range. Another patient's peak level, checked 1 hr after rFVIIa administration, was 551%, which was oversufficient (Fig. 3). If pharmacokinetic evaluation of each patient was performed before surgery, personalized factor replacement therapy and more effective coverage would be possible.
To maintain a constant concentration of FVII, continuous infusion can be more effective [17]. The use of continuous infusion as an alternative to administering rFVIIa has been investigated in hemophilia patients with inhibitors, whereas experience with continuous infusion in FVII-deficient patients seems very limited.
Efforts to maintain a target level of FVII activity have faced several problems such as the fluctuation in plasma concentrations of the coagulation factor level and the fact that measurement of FVII activity is of only limited effectiveness for monitoring rFVIIa. FVIIa assay has been reported to be better than the FVII:C assay in monitoring rFVIIa [18]. However, these findings have not been supported by clinical studies.
The patient who was asymptomatic previously underwent a dental extraction (case 6), and the procedure was performed with only antifibrinolytic agents without any replacement. The patient undergoing dental extractions was discharged from the hospital on the day of treatment. A recent study reported excellent hemostasis in 4 patients receiving rFVIIa via bolus injections for only 1 day during dental extractions [16]. The other patient had very low FVII activity at 0.6%, and rFVIIa was administered before the dental extraction. No bleeding events were observed. The patient thus underwent another dental extraction without rFVIIa; again, there was no bleeding. However, the other 2 patients, who had previous hemarthrosis and who suffered from arthropathy, underwent orthopedic surgery with rFVIIa coverage, and the results of the operations were satisfactory.
In summary, rFVIIa administration for factor VII deficiency was administered to patients with a history of frequent bleeding and had undergone major surgery. rFVIIa was first administered at 1 hr before the operation, after which it was administered every 4 hr at a dose of 15.30 µg/kg for 48 hr postoperatively; thereafter the dosing interval was adjusted according to the FVII activity and the patient's progress. FVII activity was measured at baseline and before and 30 min after the intravenous administration of rFVIIa [19].
In conclusion, rFVIIa for factor VII deficiency was well tolerated and maintained effective hemostasis with good clinical outcomes. Patients with congenital FVII deficiency who require surgery can be treated efficiently and safely using rFVIIa and antifibrinolytic agents. Knowledge of these patients' previous bleeding history and their response to rFVIIa should be helpful in managing bleeding during surgery in patients with factor VII deficiency.



 XML Download
XML Download