

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is a clinical syndrome caused by the uncontrolled activation of cytotoxic T cells and antigen-presenting cells. The initial signs and symptoms of HLH can mimic other more common conditions including bacterial sepsis, viral infections, autoimmune disease, hepatitis, and encephalitis. The early clinical signs associated with HLH include fever, hepatomegaly, splenomegaly, lymphadenopathy, rashes, and neurological abnormalities [1]. The typical histopathological findings include the widespread accumulation of lymphocytes and mature macrophages, sometimes with hemophagocytosis, especially in the spleen, lymph nodes, bone marrow, liver, and cerebrospinal fluid (CSF) [2].
HLH manifests as 1 out of 2 conditions that may be difficult to distinguish from each other: familial HLH and secondary HLH [2]. Proven mutations in genes encoding perforin-1, MUNC13-4, or syntaxin11 are diagnostic for familial HLH; a family history is also informative [3]. Central nervous system (CNS) involvement has been reported in 10-73% of all HLH patients, either at presentation or during the course of the disease. CNS involvement is highly variable in its presentation [4-6] and widely reported to be a poor prognostic factor [7, 8].
HLH is difficult to diagnose because the associated clinical symptoms mimic those characteristic of many other diseases. Since diagnostic criteria and treatment regimens with chemotherapeutic and immunomodulatory drugs have been implemented, the outcomes have significantly improved-from a 95% mortality rate previously to a cure rate of 55% [9, 10]. The early initiation of treatment is critical for enhancing a patient's chances of survival; therefore, earlier diagnosis is mandatory, and high-risk patients should undergo an intensified chemotherapy regimen.
Although the neurological manifestations of HLH have been recognized, their characteristics and impact on outcomes remain relatively poorly defined. To facilitate the identification of neurological manifestations of HLH and establish more effective treatment strategies, we reviewed the clinical data collected for 50 patients with HLH. We compared the clinical characteristics, treatment protocol, and outcomes of patients who had CNS involvement with those who did not.
MATERIALS AND METHODS
1. Patients
During the period from January 1995 to August 2011, 50 HLH cases were treated at Asan Medical Center in Seoul, Korea. Among this group, 25 patients were women and 25 men, with a median age of 2.4 years (range, 10 days to 17 years). The diagnosis of HLH required a molecular diagnosis consistent with HLH or the fulfillment of 5 or more of the following clinical diagnostic criteria:
Fever.
Splenomegaly.
Cytopenias (affecting 2 of 3 lineages in the peripheral blood): hemoglobin <9.0 g/dL, platelets <100×109/L, neutrophils <1.0×109/L.
Hypertriglyceridemia, hypofibrinogenemia, or both: fasting triglycerides ≥265 mg/dL, fibrinogen ≤1.5 g/dL.
Hemophagocytosis in bone marrow or spleen or lymph nodes without evidence of malignancy.
Low or absent natural killer cell activity.
Ferritin ≥500 mg/L.
Soluble CD25 (i.e., soluble interleukin-2 receptor) ≥2,400 U/mL [11].
A bone marrow biopsy was performed on each patient. The results showed diffuse infiltration by lymphocytes and histiocytes with hemophagocytosis in all patients. Twenty-two patients were studied with regard to genes encoding perforin-1, MUNC13-4, or syntaxin11. Patients who had neurological symptoms or elevated WBC counts in the CSF were classified as the CNS (+) group, regardless of brain imaging results or the presence of protein in the CSF.
2. Methods
We performed a retrospective chart review of patients who were treated for HLH. We reviewed the medical records for neurological symptoms and signs and current neurological states. The initial laboratory data included serological markers for hepatitis viruses (hepatitis A, hepatitis B, hepatitis C, cytomegalovirus, and Epstein-Barr virus (EBV)) and CSF findings. Computed tomography (CT) and brain magnetic resonance imaging (MRI) findings were also obtained. These clinical findings and laboratory data were described for each group, and differences were compared between the CNS (+) and CNS (-) groups.
During the study period, 3 different chemotherapy protocols were used. Immune therapy was used to treat patients diagnosed from November 1996 to February 1997. The immune therapy regimen involved combined treatment with steroids and rabbit antithymocyte globulin (ATG), followed by maintenance therapy with cyclosporine A (CSA) and intrathecal methotrexate (IT MTX), usually given every week (up to 6 doses). The number of IT MTX injections depended on the intensity of CNS disease and the CSF analysis completed before each IT injection [12].
The next group of patients (those diagnosed from September 1995 to November 2004) was treated with the HLH 94 protocol: 8 weeks of initial therapy with etoposide (VP-16) and dexamethasone. In this protocol, up to 4 doses of IT MTX were recommended for patients with progressive neurological symptoms after 2 weeks of therapy, or if any abnormal CSF condition had not improved. Maintenance therapy consisted of daily CSA plus alternating weekly pulses of VP-16 or dexamethasone [13].
The final group of patients (diagnosed from March 2005 onward) was treated using the HLH 2004 protocol, which consisted of immediate treatment with VP-16, dexamethasone, and CSA. In this protocol, the initiation of CSA treatment was moved from week 8 or later to day 1 to provide more intense immune suppression early in the course of treatment. The addition of corticosteroids to IT therapy was suggested [11].
With regard to HLH therapy status, the patients were classified as being "off-therapy" if they had been off therapy without disease re-activation for at least 1 year. Allogeneic hematopoietic stem cell transplantation (HSCT) was planned for patients with recurrent or refractory disease or with familial HLH whenever suitable donors were available.
3. Statistics
The chi-squared and Mann-Whitney U tests were used to compare data between the groups. The Cox proportional hazards regression model was used in multivariate analyses to identify the most significant prognostic factors for survival and to calculate the hazard ratio (HR) of death and 95% confidence intervals (CI) for the ratio. Survival was compared using the Kaplan-Meier life table method and analyzed by the log-rank test. All analyses were done using Statistical Package for the Social Sciences (SPSS) software, version 19. Differences were considered statistically significant if the 2-tailed P value was <0.05.
RESULTS
Fifty consecutive patients were diagnosed with HLH and then divided into 2 groups based on the presence of CNS involvement. Twenty-three patients were classified as CNS (+); the remaining 27 patients were classified as CNS (-). In the CNS (+) group, 19 patients had neurological symptoms, including seizures, facial palsy, dysarthria, dysphagia, and changes in mental status. Among these patients, 9 had elevated CSF WBC counts. Another 4 patients had elevated CSF WBC counts without neurological symptoms.
1. Clinical features of the CNS (+) group
Table 1 shows the characteristics of the 23 patients in the CNS (+) group. Four patients had CNS symptoms at the time of diagnosis. Nine patients developed CNS symptoms after diagnosis, during treatment. Thirteen patients (including 3 with febrile seizures at diagnosis) exhibited leukocytosis at the time of the initial CSF analysis (increased leukocyte count >5/µL). The mean age at diagnosis was 3.4±4.4 years (range, 10 days to 17 years). CNS symptoms included seizures (N=15), decreased consciousness (N=4), facial palsy (N=2), irritability and bulging fontanel (N=1), dysphagia and dysarthria (N=1), and brain death (N=1).
Etiologically, 5 patients had a familial history of affected siblings who died of sepsis or hematologic malignancy at <1 year of age. Another 3 were confirmed to have a UNC13D (encoding MUNC13-4) mutation, and 1 was confirmed to have a PRF1 (encoding perforin-1) mutation. With regard to infections, 6 patients had infections at diagnosis: EBV (N=5) and tuberculosis (N=1).
Twenty-one patients underwent brain CT and/or MRI examinations. Twelve patients showed abnormalities, including high signal intensity lesions on T2-weighted MRI images (N=8), ventriculomegaly (N=3), hemorrhage (N=3), atrophy (N=3), leptomeningeal enhancement (N=2), and posterior reversible encephalopathy syndrome (PRES, N=2). High signal intensity lesions with a non-specific distribution were the most common finding, and found more often in white matter than gray matter.
Neurological sequelae were reported in approximately 27.2% (3/11) of long-term survivors. Two of them took anti-epileptic drugs due to seizure disorders; another patient had facial palsy, dysphagia, and dysarthria. Among the 15 patients who presented with seizures, 2 had been taking an anti-epileptic drug (AED) and 4 had achieved seizure-free states without AEDs. The remaining 9 patients died or were lost to follow-up. On the contrary, 4 patients with only leukocytosis in the CSF exhibited normal development.
2. Clinical characteristics of patients in the CNS (+) and CNS (-) groups
There was no significant gender difference between the CNS (-) (12 men among 27 patients) and CNS (+) (13 men among 23 patients; P=0.395) groups. Patients in the CNS (+) group were younger than those in the CNS (-) group (41.0±52.8 months vs. 73.1±69.9 months; P=0.055), but this effect was not statistically significant. All 5 patients with a family history had neurological symptoms. Among the 13 patients in the CNS (+) group who were tested, 1 patient had a PRF1 mutation and 3 had a UNC13D mutation. In the CNS (-) group, 2 among 9 had a UNC13D mutation without family history. Since 10 patients in the CNS (+) group and 18 in the CNS (-) group did not agree to genetic testing, it is difficult to compare the proportion of familial HLH between the groups. With regard to infections, 18 (66.7%) patients in the CNS (-) group had viral infections (EBV (N=14), cytomegalovirus (CMV) (N=3), and hepatitis A virus (N=1)) at the time of diagnosis. In comparison, 6 patients (26.1%) in the CNS (+) group had infections at the time of diagnosis: EBV (N=5) and tuberculosis (N=1). Therefore, the rate of EBV infection was significantly less in the CNS (+) group than in the CNS (-) group (21.7% vs. 51.9%; P=0.029; Table 2).
Table 3 shows the laboratory findings of the CNS (+) and CNS (-) groups. The CNS (+) group had lower overall blood cell counts than the CNS (-) group, but this trend was not statistically significant. Other laboratory data with diagnostic value for HLH (triglyceride and fibrinogen levels) were similar, but the CNS (+) group had lower ferritin levels (P=0.037). Patients in the CNS (+) group also had significantly lower aspartate aminotransferase (AST) (P=0.026) and alanine aminotransferase (ALT) levels (P=0.011).
3. Treatment outcomes according to CNS involvement and treatment protocol
Table 4 shows the treatment protocols and treatment outcomes for each group. Three of the 23 patients with CNS disease were initially treated with immune therapy: 1 died and 2 were lost to follow-up. Out of 4 patients that began treatment on the HLH 94 protocol, 1 was alive after allogeneic HSCT and 3 died. Among 14 patients treated by HLH 2004, 7 were classified as being off-therapy, 2 were alive after allogeneic HSCT, 3 died, 1 was lost to follow-up, and 1 was receiving chemotherapy at the time of the last follow-up. The remaining 2 patients in the CNS (+) group died before starting treatment. Nine patients in CNS (+) group died due to sepsis (N=5), liver failure (N=1), brain hemorrhage (N=1), brain death (N=1), or status epilepticus (N=1).
Six patients in the CNS (-) group were initially treated according to HLH 94. Three were off therapy, 2 were alive after allogeneic HSCT, and 1 was lost to follow-up. Eighteen were treated according to the HLH 2004 protocol; 13 completed the chemotherapy regimen, 2 were alive after allogeneic HSCT, 2 were lost to follow-up, and 1 was receiving chemotherapy at the time of the last follow-up. Two died before starting treatment because of liver failure, and one was cured without treatment.
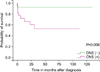
In the CNS (-) group, 2 (7.4%) patients died before starting treatment, 3 (11.1%) were lost to follow-up, and the remaining 22 (81.5%) patients were alive at the time of follow-up. Out of the 22 survivors, 4 (14.8%) had received HSCT and 16 (59.3%) were classified as off therapy. In the CNS (+) group, 9 (39.1%) patients died, 3 (13.0%) were lost to follow-up, and the remaining 11 (47.8%) were alive at the time of follow-up. Out of the 11 survivors, 3 (13.0%) had received HSCT and 7 (30.4%) reached off-therapy status. Thus, the 5-year survival rate was poor for patients in the CNS (+) group (52% vs. 92%; P=0.006; Fig. 1).
Patients treated according to the HLH 94 protocol, regardless of CNS involvement, had lower 5-year survival rate than those treated according to the HLH 2004 protocol, but this trend was not statistically significant (75% vs. 85%; P=0.443; Fig. 2A). Similarly, in the CNS (+) group, patients treated with the HLH 94 protocol had lower 5-year survival rates than those treated with the HLH 2004 protocol, but that was not statistically significant (33% vs. 67%; P=0.215; Fig. 2B). All of the treatment protocols used achieved similar rates of 5-year survival.
4. Prognostic factors influencing the survival rate
In the log-rank test, we noted that CNS involvement, age, and family history were significant prognostic factors for survival (Table 5). In a multivariate analysis using Cox proportional hazard regression analysis with backward elimination, CNS involvement was the only significant prognostic factor. The HR of CNS involvement was 5.28 (95% CI, 1.118-24.958; P=0.036).
DISCUSSION
This retrospective study of neurological manifestations of HLH suggests that about one-half (46%) of patients with HLH have neurological symptoms such as seizures or facial palsies during the course of HLH and/or abnormal CSF findings at the time of diagnosis. Janka [14] reported CSF abnormalities in 17 of a series of 33 patients and neurological symptoms in less than 10% of the patients. In a multicenter retrospective study, Arico et al. [15] reported CSF abnormalities at the time of diagnosis for 55 of 94 patients (58%). Hirst et al. [16] found neurological manifestations at the time of diagnosis in 7 of 23 patients (30%) and CSF abnormalities in 13 of 17 evaluable patients (76.5%). Henter and Elinder [17] reported CSF abnormalities in 4 of 7 patients, with neurological symptoms in 3 of them. Most published reports fail to address the frequency of CNS disease among HLH patients. In the present study, we measured the duration of CNS involvement. Fourteen patients had CNS manifestation at the time of diagnosis (3 had both neurologic symptoms and CSF leukocytosis, 1 had neurologic symptoms, and 10 had CSF leukocytosis). Nine patients developed CNS manifestations during the course of HLH. More patients had CNS manifestations at the time of diagnosis as opposed to manifestations that appeared during treatment.
Data on the incidence of neuroradiological abnormalities in HLH are limited, in particular with regard to findings at the time of diagnosis [4, 18-20]. Haddad et al. [4] reported a series of 34 patients in which they observed a variety of neurologic symptoms, including hypotonia or hypertonia, meningismus, seizures, and coma with opisthotonos. Brain MRIs of their patients showed peri-cerebral or diffuse subdural dilations and large confluent hyperintense T2-weighted signals in the cerebrum (i.e., necrotic areas). In the present study, brain CT/MRI was performed early in the course of the disease in 74% of cases; 37.8% of this group were reported as having pathological findings including high signal intensity lesions on T2-weighted MRI images, ventriculomegaly, hemorrhage, leptomeningeal enhancement, and diffuse atrophy. Interestingly, mild-to-borderline ventriculomegaly or leptomeningeal enhancement was also reported in 2 out of 27 patients without abnormal CSF findings or neurological symptoms, and 33.3% (3 out of 9) of the patients with both abnormal CSF and neurological symptoms were free of any abnormal neuroradiological findings. CNS infiltration typically begins in the meninges, and then induces perivascular changes. This leads to the diffused infiltration of tissue and multifocal necrosis at a later stage in the course of the disease [4]. It is therefore possible that neuroradiological findings may precede apparent clinical neurological manifestations. To better define the predictive value of neurological evaluation and CNS imaging, a large prospective study is needed.
When comparing the characteristics at the time of diagnosis according to CNS involvement, patients with CNS involvement had lower ferritin, AST, and ALT levels. Ferritin is involved in the regulation of iron storage and homeostasis. In addition, ferritin is a surrogate marker for inflammation because the gene promoter contains pro-inflammatory cytokine-responsive regulatory elements. Ferritin is unlikely to contribute to the pathology in HLH, but highly elevated ferritin levels may provide a weak prognostic marker. A rapid fall of ferritin levels following the initiation of therapy was reported to be associated with decreased mortality [21]. The concentration of serum ferritin at the time of diagnosis is suggested to be a prognostic factor for HLH [22]. Some studies report that the disease activity of HLH is significantly related to elevated serum levels of AST and ALT [22, 23]. However, our study showed that the CNS (+) group had a poorer prognosis despite lower ferritin, AST, and ALT levels, and there was no effect on mortality (Table 5). This result suggests that CNS involvement may be a critical prognostic factor for HLH.
Familial forms of HLH are due to genetic defects that lead to the impaired function of natural killer cells and cytotoxic T cells. Some cases of familial HLH are caused by genetic mutations that occur either in the perforin gene or in genes important for the exocytosis of cytotoxic granules. Acquired forms of HLH are encountered in association with viral infections such as herpesviruses and EBV. EBV-associated HLH has a high prevalence in Asia, suggesting the role of environmental or genetic factors [24]. There are few studies on the prevalence of infections and related family histories in HLH patients with CNS manifestations. However, a recent study of 42 HLH patients also showed that familial HLH was highly associated with poor prognoses among patients with CNS disease and/or persistently deficient natural killer (NK) cell activity [18]. In the present study, the CNS (+) group included significantly fewer patients with EBV infections. There was a strong association between CNS involvement and familial forms of HLH (P=0.014). This study showed that familial HLH was more frequently associated with CNS involvement, suggesting that genetic susceptibility plays a key role in CNS infiltration by activated lymphocytes and macrophages.
While the timely administration of chemotherapy is essential to the survival of patients with HLH, neurologic complications may be attributable to therapy and not disease in several patients treated with the HLH 2004 protocol in combination with the early administration of cyclosporine. Thompson et al. [3] reported that severe neurotoxicity (e.g., PRES) was more common among patients treated with HLH 2004 than among patients treated with HLH 94. In our study, 2 patients (Patient 19, Patient 21) treated with HLH 2004 experienced PRES, but they had CNS manifestations prior to the development of PRES. Patient 19 suffered from a seizure prior to manifesting any signs of PRES; Patient 21 had CSF leukocytosis at the time of his diagnosis with HLH. Among 9 patients who developed neurologic symptoms during treatment, 2 were treated with HLH 94 and 4 were treated with HLH 2004. Two patients treated with HLH 94 experienced their first seizure during the initial treatment before starting cyclosporine. Two patients treated with HLH 2004 showed mental changes during the initial treatment period; 2 other patients suffered from seizures and irritability during continuation therapy. Due to the small sample size, it is difficult to know the effect of the early administration of cyclosporine.
We found a significantly increased risk of mortality among patients with CNS manifestations (39.1% vs. 7.4%, P=0.006) (Fig. 1). This is in line with a recent study of 193 patients with HLH, which identified a trend towards a poorer prognosis for patients with clinical neurological manifestations and abnormal CSF findings [7]. The finding is also supported by a recent study of 48 patients with HLH, which identified a trend towards a poorer prognosis for patients with clinical neurological manifestations and abnormal neuroimaging features [8]. Imashuku et al. [18] also reported poorer outcomes in patients with CNS symptoms and/or deficient NK cell activity. The multivariate Cox proportional hazard model identified CNS involvement as the only significant prognostic factor in our study. The ability to extrapolate from this finding is limited by our study's small population size. It is difficult to enroll many pediatric HLH patients, because HLH is a rare disease. Additional studies that include more HLH patients will provide sorely needed information.
To conclude, our data indicate that CNS involvement in HLH is very frequent not only in familial HLH but also in secondary HLH; this CNS involvement is associated with poor outcomes. Nonetheless, patients with CNS involvement can recover completely with adequate therapy in spite of the progressive nature of the disease. In view of the low regenerative potential of nervous tissue, superior outcomes may require that we carefully monitor neurological manifestations in patients with HLH. Patients who have CNS manifestations of HLH will require an appropriate regimen of intensified chemotherapy. It is also important to consider HLH in a child with unexplained neurologic manifestations, especially one with fever, pancytopenia, or hepatosplenomegaly.






 XML Download
XML Download