

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Iron is a component of ribonucleotide reductase, the enzyme responsible for deoxyribonucleotides synthesis, and is known to be essential for cell proliferation and cell survival [1-3]. Evidence suggests that iron overload can cause hyperproliferation of some types of cancer cells, whereas iron depletion can inhibit proliferation in other types of cancer cells, including leukemia cells [4-8].
A number of studies have demonstrated the anticancer effects of deferoxamine, an injectable iron chelator, in some cancer cell lines, such as hepatocellular carcinoma, neuroblastoma, and ovarian carcinoma [9-11]. Deferasirox is an oral iron chelator that has been shown to have antiproliferative effects in myelodysplastic syndrome, a hematological condition characterized by ineffective production of myeloid blood cells [12]. Deferasirox inhibits nuclear factor kappa-light chain-enhancer of activated B cells (NF-κB), a transcriptional nuclear factor involved in the regulation of several fundamental cellular processes such as proliferation, differentiation, and tumor migration [13].
The NF-κB pathway can be activated by various stimuli, one of the most important being the tumor necrosis factor (TNF) receptor signaling pathway [14, 15]. TNF signaling in cells results in a subtle balance between survival and death. In fact, NF-κB activation mediated by TNF leads to an anti-apoptotic effect through both caspase and Jun N-terminal kinase (JNK) cascade inhibition [16, 17].
In 2001, Guzman et al. demonstrated that NF-κB is constitutively activated in primitive human acute myelogenous leukemia cells and suggested that leukemic stem cells are preferentially sensitive to inhibition of NF-κB [18]. It has been also reported that NF-κB blocks the mTOR pathway, resulting in decreased survival and proliferation in a myeloid leukemia cell line [19]. Another recent study also showed that deferasirox had an apoptotic effect on myeloid leukemia cells, and apoptosis was dependent on the caspase activity which is known to be associated with NF-κB [20].
Many patients with malignant lymphoma frequently received blood transfusions due to the iron overload caused by recurrent chemotherapy. While it is known that deferasirox is an iron chelator, little is understood about the cytotoxic effects of this compound on malignant lymphoma cells. The goal of this study was to define the underlying molecular signaling pathways responsible for the anticancer effects of deferasirox on malignant lymphoma cell lines.
MATERIALS AND METHODS
1. Reagent and cell cultures
The oral iron chelator deferasirox was provided by Novartis. We purchased 3 human malignant lymphoma cell lines, Ramos, Jiyoye, and NCI H28:N78, from the Korean Cell Line Bank for this study. Ramos and Jiyoye cells were incubated in RPMI 640 with 10% fetal bovine serum (FBS), and NCI H23:N78 cells were grown in DMEM with 10% FBS in an incubator at 37℃ with 5% CO2.
2. Cell proliferation assay
Cell viability was assessed by the 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide (MTT) assay. Each lymphoma cell line was seeded into 96-well plates at a 2×104 cells/well and treated with dimethyl sulfoxide (DMSO) as vehicle control or deferasirox at 20 µM, 50 µM, or 100 µM for 24 h, 48 h, or 72 h. Following treatment, cells were incubated with 50 µg/mL MTT solution (Sigma, St. Louis, MO, USA) for 4 h. Cell viability was determined by formazan formation, measured as absorbance at 595 nm by using an ELISA plate reader.
3. Cell cycle analysis
Cell cycle distribution was analyzed by flow cytometry (Beckman Coulter). Each lymphoma cell line was seeded into 60-mm dishes at 7×105 cells/dish and treated with DMSO as vehicle control or deferasirox at 20 µM, 50 µM, or 100 µM for 24 h. The cultured cells were harvested and fixed in 85% ethanol and 0.5 mM EDTA and then stained with a solution containing propidium iodide (PI; Sigma) and RNAase (0.1 mg/mL) in the dark at 4℃ for 30 min. Flow cytometry was used to determine the percentage of cells in each phase of the cell cycle.
4. Determination of apoptosis by flow cytometry
For analysis of apoptosis, cells were treated as described above. After harvesting, cells were stained with 5 µL Annexin-V conjugated to fluorescein isothiocyanate (FITC) plus 2 µL PI in the dark for 10 min. After addition of 400 µL Annexin-V binding buffer, the percentage of cells positive for Annexin-V were analyzed by flow cytometry.
5. Determination of apoptosis by luminescence assay
Each lymphoma cell line was seeded into 96-well plates at a 2×104 cells/well and treated with DMSO as vehicle control or deferasirox at 20 µM, 50 µM, or 100 µM and incubated in a 5% CO2 incubator for 24 h. The activity of caspase 3/7 was measured using Caspase-Glo 3/7 kit (Promega, Madison, WI, USA), and caspase 9 activity was measured using Caspase-Glo 9 kit (Promega), according to the manufacturers' instructions.
6. Western blot assay
Malignant lymphoma cells were cultured for 24 h following the indicated treatment. Cells were washed twice with PBS, lysed in cell lysis buffer, and sonicated. Cell debris was removed by centrifugation at 20,000×g at 4℃ for 20 min. An equal amount of protein from each sample was resolved by SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA). The membranes were stained with Ponceau S solution to confirm the uniform transfer of all samples before blocking in xxx for 2 h at room temperature. The blocked membranes were incubated with the indicated monoclonal primary antibodies at 1:1,000 for 24 h and then washed 3 times with TBS +1% Tween. Membranes were the incubated for 2 h with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (GE Healthcare, Buckinghamshire, UK). Immunoreactive proteins were visualized using ECL Western blot detection reagents and analysis system.
RESULTS
1. Cell proliferation assay
The 3 types of malignant lymphoma cell lines were treated with either DMSO (vehicle control), camptothecin (positive control), or deferasirox at 20 µM, 50 µM, or 100 µM for 24 h, 48 h, or 72 h, and then the cell survival rates were measured by MTT assay (Fig. 1). These survival rates were indicated as a percentage compared to the control (DMSO) group. After 24 h, the survival rate of the NCI cells treated with the positive control camptothecin was only 43%, which was less than the survival rate at even the highest concentration of deferasirox (Fig. 1A). The same pattern was observed for the Jiyoye cells after 24 h (Fig. 1B) and for the Ramos cells, although treatment with 100 µM deferasirox resulted in a cell survival rate slightly less than that of the positive control (Fig. 1C). All 3 cell lines showed significantly decreased survival rates depending on the concentration of deferasirox when measured after culturing for 24 h, 48 h, and 72 h. These data demonstrated that deferasirox was able to affect cell proliferation after only 24 h; therefore, the other tests were also conducted after 24 h treatment.
2. Cell cycle analysis
Cells were treated with DMSO (vehicle control) or deferasirox at 20 µM, 50 µM, or 100 µM for 24 h, and then, the cell cycle was analyzed by PI staining and flow cytometry (Fig. 2). The experiment was performed in triplicate, and data are presented as the average percentage of cells in the sub-G1 population. The measured value average of the negative control group with DMSO showed to be 2% in the NCI cell line but in deferasirox group showed 5, 9, 23% in sub-G1 portion, which verifies that the sub-G1 portion increases according to the concentration level of deferasirox (Fig. 2A). In Jiyoye cell line, the DMSO group showed 2%, deferasirox group showed 6, 9% and 13% of Sub-G1 portion depending on the level of deferasirox concentration (Fig. 2B). In Ramos cell line it showed 2, 16, 18% and 35% of Sub-G1 portion each (Fig. 2C).
3. Determination of apoptosis by flow cytometry
Early stage of apoptosis was measured using the Annexin-V Apoptosis Detection Kit (Fig. 3). The experiment was performed in triplicate, and the results are presented as the percentage of cells in the B4 portion showing early apoptosis. We found that deferasirox treatment increased the percentage of cells undergoing early apoptosis in a dose-dependent manner (Fig. 3A-C).
4. Caspase activity
Caspase 3/7 and caspase 9 activities were determined by luminescence assay (Fig. 4). In all the 3 malignant lymphoma cell lines tested, caspase 3/7, and caspase 9 activities were 2-3 fold higher in the samples treated with deferasirox compared with those treated with DMSO.
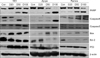
5. Western blot analysis
Protein expression of cleaved PARP, caspase 3/7, and caspase 9 was evaluated by immunoblot assay (Fig. 5). In all the 3 cell lines examined, the western blot results verified that treatment with deferasirox increased the cleavage of PARP, caspase 3/7, and caspase 9. Deferasirox treatment also increased the expression of the pro-apoptotic proteins Bax and p53 (Fig. 5). This result indicates that the anticancer effect of deferasirox was due to the activation of an intrinsic apoptosis signal pathway related to p53 and Bax.
DISCUSSION
The rate of absorption and excretion of iron in the body is maintained at a steady state. However, in the case of aplastic anemia, myelodysplastic syndrome, or other kinds of anemia, the regular cycle of blood transfusion causes iron to accumulate faster than it can be excreted. With no particular means to excrete this excess iron, it can induce damage to the organs of the body [21]. Various iron chelators have been used as therapeutic agents to remove the stored iron in the body, thereby preventing organ damage. However, the antiproliferative effects of iron chelating agents have only recently been recognized [22-24]. Deferoxamine, which is an iron chelator, has increased the survival rate of patients with iron overload [25], and some studies have also shown its anticancer effects [8, 26, 27]; however, this drug must be administered by injection rather than orally, and only 10% of patients are being treated with deferoxamine [28].
The oral iron chelator deferasirox has been recently developed and is, currently, in clinical trials, with its anticancer effects being reported [19, 29]. However, the mechanism of the drug's antiproliferative effects is yet to be determined. Chantrel-Groussard et al. reported that deferasirox can inhibit polyamine synthesis and block cell cycle in the G2-M phase by decreasing ornithine decarboxylase and spermidine N1-acethyltransferase activities as well as ornithine decarboxylase mRNA levels. They concluded that the anticancer and antiproliferative effects of deferasirox in hepatocellular carcinoma are distinct from its iron chelating activity [29, 30].
There have also been reports of deferasirox acting as a potent NF-κB inhibitor in patients with myelodysplastic syndrome [12]. Recently, Ohyashiki et al. reported that deferasirox inhibited proliferation of acute myeloid leukemia cells due to an increase in REDD1 (Regulated in development and DNA damage response) and tuberous sclerosis complex 2 (TSC2). This inhibited the phosphorylation of mTOR and decreased the phosphorylation of p70S6 kinase and S6 ribosomal protein, thus leading to inhibited proliferation of leukemic cells [19]. These findings lead us to consider that deferasirox also may have anticancer effects in malignant lymphoma cell lines.
In our study, we demonstrated that deferasirox has antiproliferative effects in human malignant lymphoma cell lines. The MTT assay showed that deferasirox induces cytotoxic effects in 3 malignant lymphoma cell lines after a 24-h treatment. Annexin/PI staining demonstrated that deferasirox induces early apoptosis in the same malignant cell lines. Western blot analysis showed an increase in the cleavage of caspase 3/7 and caspase 9 as well in the expression of P53 and Bax. To examine other cell signaling pathways, we evaluated targets in the PI3K/Akt/mTOR pathway, NF-κB signaling, and the RAS/JNK pathway, but we did not obtain significant results.
In conclusion, our study showed that deferasirox induces DNA damage in malignant lymphoma cell lines, thereby causing an increase in P53 expression and an induction in early apoptosis through Bcl2-Bax. However, the mechanism by which deferasirox causes DNA damage is not well understood. In addition, the induction of early apoptosis alone may not be sufficient to account for the anticancer effects of deferasirox. Therefore, further studies are required, perhaps, using siRNA against P53 or Bax; furthermore, in vivo animal experiments using nude mouse are needed to validate the results of the effects of deferasirox. Overall, our data suggest that deferasirox is a promising new antiproliferative agent for use in the treatment of malignant lymphoma.




 XML Download
XML Download