

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
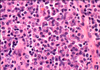
Fifty-seven years ago, Dr. Castleman [1], a pathologist at Massachusetts General Hospital, first described a rare lymphoproliferative disorder that now bears his name. Also known as angiofollicular or giant lymph node hyperplasia, the clinical manifestations of Castleman disease (CD) are heterogeneous, ranging from asymptomatic discrete lymphadenopathy to recurrent episodes of diffuse lymphadenopathy with severe systemic symptoms. The population prevalence of CD has not been established; based on the proportion of patients presenting to a large cancer centre with lymphadenopathy of undetermined origin later diagnosed with CD, it was estimated that the number of cases in the United States ranges from 30,000 to 100,000 [2]. The infrequency with which CD is diagnosed has precluded comprehensive clinical studies, leading to an incomplete understanding of the disease. Current knowledge is based largely on case series and histopathologic reviews. Over the last decade, a large body of evidence has supported the importance of infection with human herpesvirus 8 (HHV-8) or Kaposi sarcoma (KS)-associated herpesvirus in the etiology and management of CD. To date, CD exhibits several different histological patterns (Fig. 1). The hyaline vascular variant (HVV) is characterized by abnormal follicles with regressed germinal centers surrounded by widened mantle zones composed of small lymphocytes in an onion ring-like arrangement. The plasma cell variant (PCV) is characterized by hyperplastic germinal centers and a massive accumulation of polyclonal plasma cells in the interfollicular region. Marked vascular proliferation in the interfollicular region is present in both CD variants. Mixed forms demonstrate the presence of both hyaline vascular and plasma cell elements [2]. The plasmablastic variant is less common and is associated with human immunodeficiency virus (HIV) infection. A subvariant of the plasma cell form, it is characterized by large plasmablasts harboring (HHV)-8 and can progress to frank plasmablastic monoclonal lymphoma [3]. This article provides an overview of CD and its variants, proposing a classification system based on both histopathology and clinical findings. Furthermore, recent advances in the pathogenesis, natural history and treatment are summarized, emphasizing the role of HHV-8 infection.
CLASSIFICATION
Dr. Castleman initially described a patient who presented with many years of fever and weakness, and was eventually found to have a large mediastinal mass on fluoroscopy. Medical evaluation revealed mediastinal lymphadenopathy, which, on surgical excision, was found to have the strikingly abnormal architecture of the affected lymph nodes that has come to characterize CD [1]. One dozen additional patients who were largely asymptomatic but had mediastinal masses detected in the course of other medical procedures were subsequently identified [3]. The lymph nodes from these patients showed disarray in all compartments. Most striking were the lymphoid follicles, both markedly increased in number and unique in morphology. The germinal centers varied in their cellularity, ranging from a mostly acellular fibrinous hyalinization of capillaries to proliferations of pale eosinophilic cells with copious cytoplasm. The germinal center was frequently surrounded by a marginal zone of concentric lymphocytes. Increased vascularization was noted in the interfollicular space, with vessels traversing between multiple germinal centers. Finally, sinuses rarely appeared in affected lymph nodes. All of the initial patients were only mildly symptomatic, if symptomatic at all, and surgical resection resulted in cure.
In subsequent years, additional cases of patients with diffuse lymphadenopathy and the histologically characteristic CD lymph node architecture were identified [4-6]. These series also revealed a new variant of CD, which differed from the original in 2 important ways. First, the germinal centers in the involved lymph nodes differed little from normal histology and showed no evidence of hyalinization. However, concentric sheets of plasma cells surrounded the germinal centers and were prominent in the interfollicular space, which also lacked the characteristic hyper-vascularity. In addition, patients uniformly presented with a host of systemic symptoms that were uncommon in the previously described variant. This novel histological variant of CD was termed the PCV, to contrast it with the HVV that was originally described. The recognition of both patients with localized lymphadenopathy and disseminated disease led to an additional clinical categorization of CD, which is unicentric (UCD), versus, multicentric (MCD). Most recently, a third 'subvariant', known as 'plasmablastic MCD', has been described in association with particularly aggressive cases of MCD. In the first series to describe such a variant, patients with CD who had polyneuropathy, organomegaly, endocrinopathy, monoclonal proteins and skin changes (POEMS syndrome), also known as Crow-Fukase syndrome, were found to have lymph nodes that resembled those in PCV, but also had large plasma cells in the mantle zone with copious cytoplasm and prominent single or multiple nucleoli [7, 8]. A second series found the variant to be associated with HHV-8 infection and progression to plasmablastic lymphoma [9].
Because the etiology of CD has not been definitively established, it remains controversial as to whether CD variants represent different ends of the same spectrum of disease or whether they are entirely separate disease entities. The recent association of HHV-8 infection with both HVV and PCV MCD, as discussed below, and the finding of both HVV and PCV in the same lymph node argue strongly in favor of a single disease with several variants, as summarized in Table 1.
PATHOGENESIS
1. Viral stimulation
With the emergence of the HIV pandemic, there has been a resurgence of interest in CD. This followed the recognition of an association between MCD and AIDS-associated-KS, again following initial publication of case reports [10]. In 1994, Chang et al. isolated a new human gamma herpes virus from AIDS-KS lesions using representational differential analysis [11]. This virus was christened with 2 names, Kaposi's sarcoma herpes virus (KSHV) and HHV8. The virus was rapidly associated with a number of additional pathologies including classic, allograft-associated, and endemic (African) KS variants [12, 13] and with AIDS-related primary effusion lymphomas [14] and a range of post-transplant conditions such as bone marrow failure [15-17]. Even in the early 1980s, with the increasing emergence of the AIDS epidemic, clinicians recognized an association between KS and MCD, observing that the 2 diseases frequently coexisted [10, 18, 19]. Indeed, 75% of HIV-positive patients and 13% of HIV-negative patients with MCD have or will develop KS during the course of their disease [20]. The first study that linked the etiology and pathophysiology of MCD with KSHV was reported in 1995, just 1 year after the discovery of this virus. In a series of 31 patients with MCD, the virus was detected in samples from 14 of 14 HIV-positive patients including 5 patients without KS and in 7 of 17 (41%) HIV-negative patients with MCD, including one case associated with cutaneous KS. By way of controls, KSHV was detected in 1 of 51 HIV negative reactive lymph nodes and 3 of 17 HIV positive reactive lymph nodes [21]. Immunohistochemical studies demonstrated that KSHV is found in the plasmablasts within MCD lesions and these cells appear to be absent from KSHV-negative MCD [22, 23]. These plasmablasts have germ line immunoglobulin genes but are lambda light chain restricted and appear to represent pre-germinal center native B cells that are polyclonal but monotypic [24, 25]. KSHV is also present in the malignant cells of plasmablastic lymphomas which occur more frequently in patients with MCD [25, 26].
2. HIV-associated CD
In the context of HIV infection, MCD is a rare but potentially fatal lymphoproliferative disorder [20]. HHV-8 can be detected in the plasmablastic cells that characterize the disease and is considered to be a causative agent [21, 27, 28]. The clinical course of MCD is characterized by recurrent attacks, with systemic symptoms, lymphadenopathy, splenomegaly, cytopenia, and inflammation associated with high HHV-8 viral load in the peripheral blood mononuclear cells [20, 27, 29, 30]. The HIV-MCD diagnosis is based on clinical features and lymph node pathology, which is characterized by angiofollicular hyperplasia and interfollicular plasma cell infiltration [27]. HHV-8-infected cells are large cells with plasmacytic differentiation that are predominantly present within the mantle zone and can be identified using immunohistochemical staining, which reveals HHV-8-associated latent nuclear antigen-1 (LANA-1) present in these cells [22].
3. Role of IL-6
Interleukin (IL)-6 is a multifunctional cytokine produced by various cells such as T cells, B cells, monocytes, fibroblasts, and endothelial cells. It induces the differentiation and proliferation of B cells and T cells, and can stimulate hematopoietic cells. It is involved in the synthesis of acute-phase reactant proteins by the liver and is associated with the development of constitutional symptoms accompanying many inflammatory diseases [31]. IL-6 also induces the secretion of the peptide hormone hepcidin by the liver, which interferes negatively with the absorption and use of iron, leading to the anemia of chronic disease [32]. Recently, its role in the growth of tumor cells and angiogenesis has suggested that it is a potential therapeutic target in cancer [33]. The contribution of IL-6 to the pathogenesis of CD has been demonstrated in animal models. Retroviral transduction of the coding sequence of murine IL-6 into the bone marrow of mice resulted in the development of a CD-like syndrome with fever, anemia, leukocytosis, hypoalbuminemia, polyclonal hypergammaglobulinemia, marked splenomegaly, and diffuse lymphadenopathy [34]. Katsume et al. [35] studied the effect of blocking IL-6 in transgenic mice carrying human IL-6 DNA using an anti-IL-6 receptor (IL-6R) monoclonal antibody. Control mice developed plasmacytosis, mesangial proliferative glomerulonephritis, leukocytosis, thrombocytosis, and anemia. Mice that received the monoclonal antibody did not develop these symptoms and lived longer. Yoshizaki et al. [36] noted an elevated concentration of IL-6 in a patient with UCD, which normalized after lymph node excision along with the disappearance of clinical and biologic abnormalities. The detection of IL-6 in germinal center B cells implicates the generation of IL-6 by B cells in the pathogenesis of MCD.
4. Role of angiogenesis and vascular endothelial growth factor (VEGF)
Blood vessel proliferation is an important histological component of CD, although the role of angiogenesis in the pathophysiology of CD has received scant attention. Nishi et al. [37] found the level of VEGF in sera and supernatants of cultured lymph nodes to be higher in 2 patients with CD than in normal controls. Immunohistochemical analysis showed a high expression level of VEGF in plasma cells of the interfollicular region [37]. In contrast, Foss et al. [38] showed overexpression of VEGF mRNA in nonlymphoid cells in patients with MCD, whereas Cohen et al. [39] demonstrated the role of IL-6 in the production of VEGF in vitro.
PATHOLOGY
The various types of CD are characterized by distinctive lymphoid architectural changes in all nodal compartments. Types of UCD include HVV and PCV. MCD is predominantly of the PCV with a few cases showing plasmablastic characteristics.
1. Hyaline vascular variant
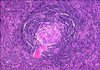
In hyaline vascular CD, involved lymph node follicles have widened mantle zones made of concentric rings of small lymphocytes (in an "onion skin" pattern) that surround small atrophic or "regressively transformed" germinal centers (Fig. 2). The germinal centers often have penetrating small hyalinized vessels and prominent follicular dendritic cells which may be expanded, be disrupted or have multiple tight connections [40]. Interestingly, the degree of follicular dendritic cell dysplasia also correlates with expression of epidermal growth factor receptor. The upregulation of epidermal growth factor receptor in the follicular dendritic cells of CD is paralleled by an increase in epidermal growth factor receptor expression in the surrounding perifollicular fibroblastic reticulum cells [41]. The interfollicular region is composed of small T lymphocytes and vascular proliferation including many high endothelial venules. Plasmacytoid dendritic cells are also present in the interfollicular region and produce large amounts of interferon-α after stimulation [42].
2. Plasma cell variant
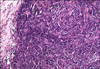
The germinal centers in the PCV CD are hyperplastic instead of regressively transformed and the interfollicular region of the node is vascular and contains sheets of plasma cells that are characteristically polyclonal (Fig. 3). Sinuses may be patent. These histopathologic findings are not specific for CD and disorders that can cause similar changes in hyperplastic reactive lymph nodes, such as rheumatoid arthritis and viral lymphadenitis, need to be excluded [4, 43].
4. Plasmablastic variant
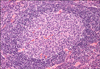
The plasmablastic variant of CD is seen most commonly in association with HIV infection and is characterized by the presence of large plasmablasts (immunoblasts) in the mantle zone that express the HHV-8 latent nuclear antigen (Fig. 5). These plasmablasts also express high levels of lambda light-chain restricted IgM, but are polyclonal and do not contain somatic mutations in their IgV genes, indicating their origin from naive B cells [25]. They may coalesce to form "microlymphomas", which can progress to frank plasmablastic monoclonal lymphoma and are clinically very aggressive [24].
CLINICAL FEATURES
The clinical findings and course differ greatly among the various types of CD. UCD hyaline vascular CD is the most common type, accounting for approximately 70% of patients with CD, and presents equally in men or women. A single node or chain of lymph nodes is involved, usually in the mediastinum, but may also occur in the cervical, axillary or abdominal regions. The enlarged nodes may be identified either incidentally or through symptoms related to local mass effects. Systemic symptoms are unusual [45]. In the UCD PCV, however, constitutional symptoms and laboratory abnormalities, much like in MCD, may be prominent in addition to enlarged lymph nodes. This subtype represents about 10-20% of cases [4].
Although MCD is less common, it is far more varied in clinical manifestation than UCD. Systemic symptoms, sometimes severe and life-threatening, are the presenting feature in most cases. The great majority of patients experience fevers, which may be quite high, night sweats, weakness, fatigue, anorexia and significant weight loss. They may become edematous and develop ascites, pleural or pericardial effusions and skin rashes. Occasionally, frank central nervous system symptoms, such as seizures, may occur. Virtually all patients have multifocal lymphadenopathy and hepatosplenomegaly is found in more than of half the cases. If MCD is suspected on clinical grounds, a generous biopsy, preferably an excisional biopsy is required. Even with characteristic histopathologic findings, other conditions, such as rheumatologic conditions and viral infections, should be excluded. The evaluation of a patient with an established diagnosis of MCD will likely show a wide range of laboratory abnormalities. Anemia, especially anemia of chronic disease or hemolysis, is present in a majority of patients along with frequent thrombocytopenia, elevated erythrocyte sedimentation rate, hypoalbuminemia, abnormal liver function test results and a polyclonal increase in immunoglobulins [46]. The patient may exhibit renal failure, including nephritis. Excessive IL-6 production can replicate many of these manifestations of CD.
The clinical course is variable and may be progressive over months or episodic with recurrent exacerbations over years [47]. Occasionally, patients may be essentially asymptomatic or have spontaneous abatement of symptoms.
ASSOCIATED CONDITIONS
A variety of related processes and other diseases have been associated with CD. Some of these associated conditions result from the immune perturbation that may occur with CD or, perhaps, forerunners of its evolution. Other conditions may be direct manifestations of the disease.
1. HIV-associated CD
The frequent observation of MCD in patients with HIV may be related to HHV-8 coinfection or, possibly, HIV may itself predispose to development of CD [48]. HIV-associated CD is mostly plasma cell or plasmablastic. Symptoms may be severe and flares of the disease can occur at any CD4 count [20]. HIV-associated MCD generally has a rapidly progressive clinical course [24]. A median survival of 14 months was noted in a relatively large series of MCD in HIV-positive patients [20]. Interestingly, rapid progression of MCD has been reported after initiation of highly active antiretroviral therapy (HAART) for HIV infection [49], but this finding is inconsistent between patients [50]. HIV-positive patients are also usually coinfected with HHV-8, which may contribute to their poor outcome.
2. Kaposi sarcoma (KS) and CD
Two types of recurrent malignancies, lymphoma and KS have been described to occur during the course of MCD in 18% and 13% of cases, respectively 46. Both the cutaneous and nodal forms of KS have been reported to be associated with this disease [18, 19, 44, 47, 51-54]. Similar clinical and pathologic features of MCD have been found in HIV-infected patients with lymph node hyperplasia [10, 55, 56]. Striking evidence is the close association between KS and MCD in these HIV-associated cases, with the presence at diagnosis or the subsequent development of KS found in 75% of patients with MCD [21]. Sequences of a putative new herpesvirus (descriptively named KSHV) have been isolated in patients with MCD by representational difference analysis [11]. KSHV sequences share homologies with Epstein-Barr virus (EBV) and herpes virus sequences and have been detected in almost all of the AIDS-associated KS samples of both forms, cutaneous or nodular [11, 12, 57-61]. Moreover, KSHV sequences have also been detected in non-AIDS KS, classic or endemic (central Africa), whereas KSHV has rarely or not been detected in panels of non-KS controls [11, 12, 57-61] with the exception of a rare form of lymphoma occurring in HIV-infected patients (AIDS-related body-cavity-based lymphomas) [14]. Infection with the putative new Herpesvirus-like agent appears to be a major factor in the development of KS.
3. Non-Hodgkin lymphoma
Non-Hodgkin lymphoma (NHL) is associated with MCD (18% in one series) [46], and the association among HIV, MCD and NHL is especially strong. Hodgkin lymphoma has also been reported to be associated with CD. Lymphoma and CD coexist at times, but the clinical course of MCD is such that it may progress to lymphoma. In a prospective study of 60 patients with HIV and MCD who were followed up for a median of 20 months, 23% developed NHL, at least half of which had aggressive plasmablastic lymphoma. The HIV viral load or CD4 count was not predictive of the risk of developing NHL [26].
4. POEMS syndrome
POEMS syndrome is a very rare form of plasma cell proliferation with lymph node features similar to those of the PCV of CD. MCD is frequently associated with POEMS syndrome and has been proposed as a minor criterion in its diagnosis [62]. HHV-8 may be seen in POEMS syndrome and its presence is predominantly seen in the subset with MCD [63]. Elevated IL-6 levels have also been documented in patients with POEMS syndrome [64].
RADIOGRAPHIC APPEARANCES
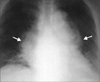
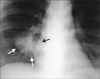
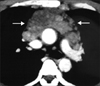
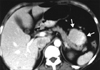
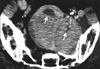
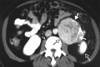
Typical thoracic CD usually occurs in the mediastinum and hilum and manifests as a rounded solitary mediastinal or hilar mass in asymptomatic patients [4, 65, 66]. On chest radiographs, mediastinal CD may mimic thymoma, lymphoma, or neurogenic tumor (Fig. 6), although hilar CD may simulate bronchial adenomas [65]. Uncommonly, thoracic CD arises from other locations, including the pleura, pericardium, intercostal space, and lung, with atypical imaging features. Pleural CD may present as either a well-defined interlobar mass (Fig. 7) or massive pleural effusion [67]. Pericardial CD may present as a pericardial mass resembling a pericardial cyst [68]. Intercostal CD may manifest as an extrapulmonary mass with rib erosion [69]. Intrapulmonary CD may appear as a solitary lung mass [4] (Fig. 8). Most instances of abdominal or pelvic CD may not be visible on abdominal radiographs unless they are massive or calcified. It is unusual for cases of CD to have calcification sufficient to be visible on radiographs, but when present, the calcification is characteristically coarse or branchlike [4, 65, 66]. Conversely, MCD may manifest as bilateral hilar and mediastinal enlargement, diffuse reticulonodular pulmonary infiltrations, hepatosplenomegaly, and ascites [66].
1. Appearance on CT
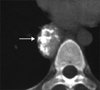
On the basis of computed tomography (CT), thoracic UCD can be categorized as 1 of 3 morphologic patterns: a solitary, noninvasive mass (50% of cases) (Fig. 9); a dominant mass with involvement of contiguous structures (40% of cases); or a matted lymphadenopathy confined to a single mediastinal compartment (10% of cases) [65, 66] (Fig. 10). Homogeneously intense contrast enhancement, reflecting hypervascularity of the lesion, is considered to be the CT finding characteristic of thoracic and abdominal UCD (Fig. 11). Prominent feeding vessels may occasionally be shown. However, Meador and McLarney [70] have reported that in the presence of intralesional fibrosis, necrosis, and degeneration, abdominal or pelvic CD may have a heterogeneous appearance on CT, especially lesions larger than 5 cm [70, 71] (Fig. 12). A few instances of mediastinal CD may also show intratumoral hypodensity on contrast-enhanced CT. On CT, 5~10% of cases of CD showed intralesional calcifications, typically being discrete, coarse, or distinctive with an "arborizing" pattern in morphology [65, 66] (Fig. 9 and 13). Intrathoracic MCD typically exhibits bilateral hilar and mediastinal lymphadenopathy, centrilobular nodular opacities, and, less commonly, ground-glass attenuation, air-space consolidation, and bronchiectasis [66]. In the abdomen, MCD is characterized by diffuse lymphadenopathy, hepatomegaly, splenomegaly, ascites, and thickening of the retroperitoneal fascia [70].
TREATMENT
Early interventions in the treatment of CD were iterations of standard therapy for lymphoproliferative diseases, including surgical excision, cytoreductive chemotherapy and radiation therapy (Fig. 14). With a greater understanding of the pathogenesis of CD, therapies directed at specific targets have been developed and show great promise. Surgical excision case series have repeatedly illustrated that surgery is almost always curative for UCD of either the HVV or PCV [3, 4, 72, 73]. Because of the disseminated nature of the lymphadenopathy with multicentric disease, complete surgical debulking is rarely possible. The few patients with MCD in whom surgery has been reported have not had meaningful or lasting improvements in their disease symptoms [72, 73]. Because splenomegaly is a constant feature of MCD, diagnostic or therapeutic splenectomy is often performed but has not been shown to be of clinical benefit.
1. Surgical excision or localized radiation
Dr. Castleman noted in his original papers that surgery could be curative for localized CD [1, 3]. This has subsequently been confirmed. Surgical removal is almost always successful for UCD, whether HVV or PCV [72], and may be applicable even in the abdomen [74]. When surgical resection is not a good option for localized disease, irradiation is an effective alternative, with response rates up to 72% [75]. Occasionally, in symptomatic patients with MCD, the surgical removal of involved lymph nodes or spleen may cause an improvement in systemic symptoms, probably related to a "debulking" effect, but any improvement is usually transient [47]. Unlike patients with UCD, the symptomatic patient with MCD requires systemic therapy and the several available alternatives should be matched to the patient's needs and overall condition. The rare asymptomatic patient with MCD is probably best observed without specific intervention.
2. Corticosteroids
The use of prednisone or other glucocorticosteroids will frequently promptly ameliorate symptoms, partially improve lymphadenopathy and correct laboratory abnormalities. The impact is generally temporary, however, and symptoms are likely to recur upon tapering or discontinuing the treatment. Lasting remissions are rare and the disease may require the long-term use of corticosteroid, increasing the risk of bacterial infections that may be fatal [47, 76]. The use of corticosteroids, alone, may best be reserved as a temporary intervention in acute situations where more definitive therapy has not yet been decided or will be delayed.
3. Chemotherapy
Owing to the progressive nature of the findings in most patients with MCD, a lymphoproliferative process, it is not surprising that the varieties of chemotherapy used in the treatment of NHL have been utilized. Currently, chemotherapy will be the first option chosen in most symptomatic patients. There are, however, insufficient data to favor one program for the treatment of all patients. Often the choice is based on the apparent aggressiveness of the disease. Chemotherapy ranging from the use of single agents to multidrug combinations has been successful and has also failed. Oral chlorambucil and cyclophosphamide have been effective in some patients, and are generally well tolerated [46, 47, 75]. Single-agent vinblastine [20], oral etoposide [77] and interferon [78] may also have activity. Single alkylating agent therapy may be most appropriate for the patient who is fragile or in whom a prompt response is not required. Combination chemotherapy regimens that have established roles in NHL, such as cyclophosphamide, vincristine and prednisone (CVP) or cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP), have significant activity [46, 47, 72]. When combination chemotherapy is used, patients need to be carefully selected and closely monitored because of the elevated risk of infection. Patients with HIV-associated CD may be at especially high risk for complications with standard combination chemotherapy. Although it is likely that the tolerance of HIV-infected patients would be improved through the use of HAART, potential drug interactions need to be considered [79]. In addition, it has been suggested that the use of HAART may precipitate or exacerbate MCD [50].
4. Radiation therapy
Radiotherapy does have therapeutic activity and is a treatment option for patients not deemed good surgical candidates or in patients with incomplete surgical excision (Table 2). The review of the literature documents that radiotherapy has the ability to achieve complete radiographic and clinical resolution of disease in patients with UCD [80]. Chronowski et al. described 4 patients with UCD treated exclusively with radiotherapy [75]. Two fractionation schedules were used: 2 Gy per day to a total dose of 40 Gy and 1.8 Gy per day to a total dose of 39.6 Gy. The median follow-up was 30 months. Two patients were alive and clinically free of disease. Two patients died with no evidence of disease at last follow-up. Three patients had complete, radiographic resolution of their lymphadenopathy. Other studies have described 8 patients with UCD treated with radiotherapy [81-87]. Three of the 8 patients had complete resolution of their lymphadenopathy, 2 patients were rendered asymptomatic but had persistence of lymphadenopathy, 1 patient had a decrease in the size of his tumor, 1 patient had a minimal response, and 1 patient had disease recurrence after initial resolution. Patients were treated with doses ranging from 27 to 45 Gy.
The study by Neuhof and Debus [88] emphasizes radiotherapy as a successful treatment option for patients with UCD. Two patients treated with surgery (partial resection) and postoperative radiotherapy showed no progression of disease during follow-up: one patient was in complete remission and the other patient in partial remission. Among the 3 patients treated with radiotherapy alone 2 patients showed no progression of disease: 1 patient had stable disease and the other patient was in complete remission. The patient with complete remission had no evidence of disease for about 14.5 years. Only one patient treated with radiotherapy alone showed progressive disease and died of CD 5 months after radiotherapy. This patient differed in clinical and tumor characteristics from the other patients; the patient was older (71 years), in bad general condition at the beginning of radiotherapy, and had bone metastases of a prostate cancer that were irradiated 9 years before the diagnosis of CD. The other patients had all circumscribed tumors in the affected lymph nodes. It seems that radiotherapy of CD has limited therapeutic activity in cases with diffusely infiltrating tumors. In the study by Neuhof and Debus one patient treated with radiotherapy alone showed serious acute and late toxicities. At the end of radiotherapy, dermatitis (grade 3) of the irradiated skin and a paraneoplastic pemphigus vulgaris (PNP) occurred. Patients with CD and PNP have a high mortality rate that is due to the almost uniform finding of pulmonary involvement leading to respiratory failure and death. In a retrospective study including 28 patients with CD and PNP, 22 patients (79%) died from respiratory failure [89]. A review of the literature documents no case of stenosis of the esophagus or trachea after radiotherapy of CD. A review of 18 cases of CD that were treated with radiotherapy found that 13 patients (72%) had a complete or partial response to radiotherapy [75]. Only 3 of the 13 responders (23%) had multicentric disease; the benefit in these patients may have been attributable to adjuvant chemotherapy or corticosteroids that were concurrently administered. Table 2 summarizes the studies that discussed the role of irradiation in the treatment of CD.
5. Immune modulators
1) Interferon-α
Significant clinical benefits have been described from single agent interferon-α for treating patients with CD. It was used initially in HIV-positive patients, but later was found to be beneficial for HIV-negative patients. One case complicated by pancytopenia required treatment interruption [90], even though interferon was well tolerated in most cases. Interestingly, interferon-α was successful in yielding long-term complete remission in patients off treatment for as long as 4 years [91]. The precise underlying mechanisms of these salutary outcomes have not been fully characterized but could be a result of several diverse biologic effects of interferon, including inhibition of trans-signaling via downregulation of IL-6R, antiviral effects such as inhibition of HHV-8 replication, and upregulation of HLA class I expression on HHV-8-infected cells leading to cell-mediated destruction [78, 92].
2) All-trans retinoic acid
All-trans retinoic acid has been shown to have antiproliferative effects [93] and may also decrease IL-6-dependent cell signaling [94]. It was hypothesized that both these properties could be beneficial in the treatment of MCD, and a case report describing its successful administration in an HIV and HHV-8 uninfected woman has been described [95].
3) Thalidomide
Similar to interferon-α and all-trans retinoic acid, thalidomide also has immunomodulatory properties [96]. Thalidomide, however, may act specifically to decrease the production of IL-6, but also possess anti-angiogenic properties. Two patients have been reported to receive thalidomide. One HIV- and HHV-8 infected man had improvements in platelet count but persistent constitutional symptoms with thalidomide and etoposide [97], and one HIV-negative woman (HHV-8 infection status not stated) had a complete response lasting over 1 year with 300 mg of thalidomide daily [98].
4) Monoclonal antibodies (anti-IL-6 & anti-IL 6R antibodies)
In recent years, the promising preclinical and clinical efficacy exhibited by targeting IL-6 or IL-6R has confirmed IL-6 as an important target in the treatment of CD. Initial evidence was examined by Beck et al. [99], who reported a case of MCD associated with elevated IL-6 levels and treated with BE-8, a murine anti-IL-6 monoclonal antibody. All clinical and laboratory abnormalities improved rapidly after initiation of treatment. However, the disease relapsed after termination of treatment [99]. The short half-life of the murine monoclonal antibody and its neutralization by human anti-mouse antibody could explain why murine monoclonal antibodies produced only a transient response. To overcome these limitations, humanized and chimeric monoclonal antibodies with longer half-lives and a lesser degree of immunogenicity were later developed. Immediate symptom relief and improvement in biochemical abnormalities were seen with the use of the humanized anti-IL-6R rhPM-1 in 7 patients with CD, 3 of them had amyloidosis. Treatment was well tolerated with only transient leukopenia. However, the disease flared up right after discontinuation of treatment [100]. In another trial conducted by Nishimoto [101], tocilizumab, a humanized anti-IL-6R monoclonal antibody, was studied in 28 patients with HIV-negative CD. Reversal of inflammatory parameters, alleviation of constitutional symptoms, and reduction in the degree of lymphadenopathy were observed. Treatment was well tolerated, with only some minor to moderate reactions, and 27 patients (96.4%) continued to receive treatment with tocilizumab for >3 years. Of 15 patients taking corticosteroids at the initiation of treatment, 11 were able to reduce the dose of or discontinue corticosteroid treatment [101]. This molecule is approved in Japan for CD. Another anti-IL-6-based therapy that has been attempted is siltuximab, a chimeric murine monoclonal antibody neutralizing IL-6. Interim results from a phase1 trial with siltuximab in patients with HIV-negative HHV-8-negative CD are available from 23 patients, all but one of whom had MCD [102]. None of those patients had drug-limiting toxicity and the treatment was well tolerated at a dose of up to 12 mg/kg weekly. Eighteen of the 23 patients (78%) achieved a clinical benefit response. Objective tumor responses were seen in 12 patients (52%). In the subgroup of patients treated at the 12-mg/kg dose level every 1, 2, or 3 weeks, 8 of 12 patients achieved an objective tumor response (73%). A separate report described a striking complete response in a patient with a refractory cutaneous form of CD after receiving 6 doses of CNTO-328 [103].
6. Antiviral therapy
Several antiviral medications have shown the ability to effectively inhibit the replication of HHV-8 in vitro, including ganciclovir, foscarnet and cidofovir [104-106]. A handful of patients with HHV-8-associated MCD have been treated with antiviral medications, with mixed success. Of 4 patients treated with foscarnet, 2 had no improvement [107, 108] and 2 had persistent remission for 12-24 months [109, 110]. Seven patients treated with a combination of cidofovir and chemotherapy exhibited no response in either symptomatology or HHV-8 viremia [108, 111, 112]. The use of ganciclovir or the oral derivative valganciclovir, however, reduced HHV-8 viremia in 3 patients and led to remissions lasting 12-18 months [113]. A number of factors may explain the heterogeneity in the response to antiviral therapy for HHV-8-associated MCD. First, because the current in vitro models may not adequately characterize the in vivo efficacy of these drugs, clinical studies to determine which of the antiviral medications has the greatest effect on HHV-8 are needed. Next, if the symptoms of HHV-8-associated MCD are attributable, at least in part to the production of vIL-6, then the current medications that inhibit DNA synthesis may fail to uniformly abort the production of this early-lytic gene product [114]. Finally, the optimal time to administer antivirals is not currently understood. If the relationship between MCD and HHV-8 is akin to that of post-transplant lymphoproliferative disorder and EBV, then the use of antivirals before cell transformation may be a successful strategy.
7. Highly active antiretroviral therapy
The implementation of HAART to treat patients with HIV/AIDS has altered the natural history of HIV and dramatically boosted survival. In patients with HIV and MCD, exacerbation of MCD during the initiation of HAART has been reported and attributed to immune reconstitution syndrome [50]. Nevertheless, the overall survival rate is remarkably higher and the progression rate to lymphoma has been 10-20 times lower in HIV-related MCD since the introduction of HAART [26, 49, 115]. Whether this is a result of HAART or the use of new chemotherapy for CD remains unclear.
8. Rituximab
Rituximab therapy seems to be a promising first-line treatment for HIV-associated MCD: In the study by Bower et al. [116] patients completing 4 weekly infusions achieved clinical and biochemical remission within 1 month, and the radiologic response rate was 67%. Plasma KSHV viral load significantly decreased in individuals with this measurement (P=0.018). The 2-year overall survival rate was 95% (confidence interval, 86-100%), and the relapse-free survival rate was 79% (confidence interval, 52-100%). This compares favorably with the median survival of 14 months recorded for 20 patients from the pre-HAART era [20]. The clinical response to rituximab occurred within 1 month of completing therapy, and normalization of acute-phase inflammatory markers, such as erythrocyte sedimentation rate, C-reactive protein, and albumin, occurred by this point. Plasma KSHV DNA viral load decreased dramatically with treatment and increased at relapse. The high plasma titers of KSHV reflect lytic replication, which is not a feature of KS but correlates with disease activity in MCD. KSHV-infected B-lymphocytes from lymph nodes in patients with MCD are known to express KSHV lytic gene products [22, 117]. Rituximab produces a decrease in CD19-positive B-lymphocytes, as would be expected, and is well tolerated in patients with HIV-related MCD, with no grade 3 or 4 toxicities. In addition, rituximab does not seem to cause exacerbation of HIV infection, with no adverse effect on the immune T-cell subsets, including CD4 cell count or HIV viral load. However, KS may progress with this treatment, a phenomenon that has been recorded previously [118]. The reason for this is unclear, but the rapid decrease in B-lymphocytes observed with rituximab therapy may play a role in the progression of KS [119]. A recent trial in patients with chemotherapy-dependent, HIV-associated MCD reported that infusion of rituximab 4 times weekly resulted in sustained remission off treatment at day 60 (the primary end point) in 92% of individuals [120]. Again, exacerbation of KS was the most frequent side effect.
9. Treatment of HIV-associated CD
The approaches used for the HIV-positive population are slightly different from the approaches for those not infected with this virus. Because these patients are already severely immunosuppressed, high-dose combination chemotherapy is a riskier prospect. Unlike the dramatic improvements in active KS after the institution of HAART, MCD does not regress by mere immune reconstitution [49, 107] with only a few exceptions [121]. There was one report, which has not been substantiated, that HAART might aggravate the symptoms of MCD [50]. The use of HHV-8 directed antivirals, such as ganciclovir, foscarnet, and cidofovir in HHV-8-positive patients has yielded conflicting results, with the majority of cases suggesting no benefit [111, 112].
Singly or in combination, liposomal doxorubicin, oral etoposide, and vinblastine have produced remission-sometimes durable-in HIV-positive patients [20, 49, 77, 122]. Vincristine, bleomycin and vinblastine combinations have been used with success [122]. Alkylator-based treatments including low-dose chlorambucil have occasionally been helpful [122]. More intensive alkylator-based therapies can result in responses, but they should be used with caution because of their extreme immunosuppressive effects [123]. Interferon-α has also provided modest benefit [78, 110]. There is one case report of the benefit of thalidomide in an HIV-positive patient with MCD [97]. Twelve patients with HIV-associated CD have been treated with rituximab with mixed results [111, 118, 124]. In the largest series, Marcelin et al. [118] reported on 5 patients infected with HIV who had CD [118]. Two died very quickly after the beginning of rituximab therapy and 3 had complete remission with no clinical symptoms related to CD with a follow-up of 4-14 months. In 2 of the responders, clinical remission correlated with a dramatic decrease of HHV-8 viral load as well as a transitory but sharp decrease of CD19 cell count and an aggravation of KS [118].



 XML Download
XML Download