

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Although the incidence of acquired hemophilia A (HA) is low in the general population, i.e., approximately 1-4 cases per million annually, it is a potentially life-threatening bleeding disorder. HA is characterized by the development of autoantibodies directed against plasma clotting factors, most frequently coagulation factor VIII (FVIII) [1, 2]. The development of autoantibodies leads to FVIII deficiency, which results in insufficient generation of thrombin [3]. Acquired HA is one of several autoimmune diseases, in which patients previously experienced normal hemostasis [4]. The age distribution of patients with acquired HA follows a biphasic trend, with a minor peak in young postpartum women and a major peak at 60-80 years in patients with other autoimmune disorders [2]. Although acquired HA is thought to be related to dysfunction of the immune system, the cause of this disorder remains unclear.
The F8 genotype is important in the molecular diagnosis of HA as well as for the prediction of risk factors pertaining to the development of inhibitory autoantibody. Mutations associated with the absence of the gene product lead to a phenotype indicative of severe HA. Patients with this disorder are more prone to developing inhibitors. Oldenburg and Pavlova [5] have previously described the relationship between mutations and inhibitor development: multi-domain large deletions pose an extremely high risk (88%) for the development of inhibitors, and single-domain large deletions, nonsense mutations, intron 22 inversions, and small deletions have risk probabilities of 25%, 31%, 21%, and 16%, respectively. However, the F8 genotype in patients with acquired HA has not been studied in detail. In addition, the genotype database for acquired HA is not sufficient to understand the correlation between mutation type and autoantibody development. Therefore, profiling of mutation types by F8 genotyping is necessary.
CASE REPORT
We examined the F8 genotype of 2 Korean patients with autoantibodies; one patient was a 71-year-old man (AMC-HA51), and the other was a 26-year-old woman (AMC-HA52). The 71-year-old man was transferred from Seoul St. Mary's Hospital for the treatment of a bladder hematoma. He was treated with cryoprecipitate in Seoul St. Mary's Hospital. Laboratory tests revealed that aPTT was 46 seconds, circulating FVIII level was 14.2%, and FVIII inhibitor was detected at 1.0 Bethesda units/mL. He was treated with steroids and cyclophosphamide for 6 months. After ceasing all medications for acquired hemophilia, tests revealed that FVIII and inhibitor levels were normal. The 26-year-old woman presented with a 3-week history of right hip pain. Her laboratory data showed that aPTT was above 200 seconds, and this was not corrected by normal FFP in a mixing test. Factor assay showed that circulating FVIII was 1.8% of the normal level and FVIII inhibitor was detected at 110.2 Bethesda units/mL. In addition, FIX activity was 45.1% of the normal levels and FIX inhibitor was detected at 0.4 Bethesda units/mL. Anti-nuclear antibody (ANA), hepatitis B-virus (HBV), hepatitis C-virus (HCV), and human immunodeficiency virus (HIV panels were negative and neither lupus anticoagulant nor anti-cardiolipin antibody was significantly detected. To control acute bleeding, we started treatment with activated prothrombin complex concentrates (aPCC) (FEIBA) at 5,500 units, twice per day (100 IU/kg every 12 h). For inhibitor suppression, the immune suppressants cyclophosphamide (50 mg/day) and prednisone (2 mg/kg) were administered. The level of inhibitor decreased gradually and improvement in FVIII activity followed. After medication had been stopped, the inhibitor remained was undetectable.
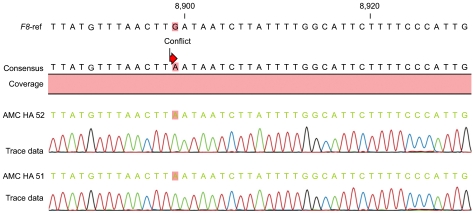
To profile the F8 genotype of these patients, we performed a direct sequencing analysis of all 26 exons, including the 3'-untranslated region (UTR) and 5'-UTR, using 35 primers, which we have previously reported [6]. Genomic DNA was purified from the whole blood of 2 patients. Tests for inversion mutations in intron 22 and intron 1, as well as gross exon deletion tests, were excluded because these mutations are known to cause congenital hemophilia or episodes of severe bleeding. A wild-type F8 gene sequence (accession no. NM_000132.3) served as the reference sequence. Finally, the mutation data were confirmed by using the CLC workbench program. Sequencing results revealed that the patients shared a common point mutation (c.8899G>A; Fig. 1) in the 3'-UTR of exon 26.
DISCUSSION
In this study, we found that 2 Korean acquired HA patients shared a common point mutation, c.8899G>A, in the 3'-UTR of exon 26 of the F8 gene. This mutation is novel and has never been reported in the HAMSTeRS database. In a previous study, it was reported that some variants of the F8 gene are related to non-congenital HA development [7]. The missense mutation, c.6238G>A, caused a substitution in the A3 domain (E2004K). A second polymorphism, c.3951C>G, caused a further substitution in the B domain (D1241E). These 2 sequence variations are related to a human leukocyte antigen (HLA)-DRB1 genotype [7]. Thus, major histocompatibility complex (MHC) background could be a risk factor for inhibitor formation in non-congenital hemophilia. In addition, cytotoxic T lymphocyte antigen 4 (CTLA4) has been reported to play an important role in the immune response [8]. Among the reported polymorphisms (-318C/T, 49A/G, and CT60 G/A), only the G allele of 49A/G has been detected at a significantly higher frequency in acquired HA patients [8-11]. The above-mentioned high frequency of HLA class II alleles and SNPs in the CTLA4 gene have been observed in acquired HA patients.
In contrast to these mutations, which occurred in coding regions, the mutation found in this study is located in the 3'-UTR region. Thus, we considered the possibility that this mutation is related to susceptibility to developing acquired HA. It has previously been reported that sequence variation in the 3'-UTR is associated with transcriptional modifications, splicing, or mRNA stability [12, 13]. In 5 families with hemophilia B (HB), a point mutation in the 3'-UTR of the F9 gene greatly reduced plasma FIX concentration to less than 3% of the normal level [14]. In addition, a 653-bp deletion located in the 3'-UTR of the F9 gene was found to be responsible for mild HB [15]. Although these reports concern the F9 gene, we suggest that the novel mutation in the 3'-UTR of the F8 gene discovered in cases of acquired HA may also be related to F8 mRNA stability or translational capacity. We surmise that an alteration in the stability of F8 mRNA and/or translational modifications leads to a synergetic triggering effect on antigen recognition between exogenous FVIII and endogenous FVIII.
Although the number of patients in this study is too few to make any important conclusions regarding the cause of acquired HA, we expect that the novel mutation discovered in this study may contribute to our understanding of the cause(s) of acquired HA. To the best of our knowledge, this is the first report on the genotyping of F8 in the Korean patients with acquired HA. Additional genotyping data from acquired HA patients will help us to determine more precise correlations between F8 genotype and the development of autoantibodies.
 XML Download
XML Download