

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Clinical laboratory diagnosis is essential for therapeutic intervention in most diseases [1]. In this context, it is important to exclude interference by other conditions that may give false positive or false negative results, leading to inappropriate diagnosis [1, 2].
Thalassemia is the most common genetic disorder worldwide [3, 4]. To date, more than 200 causative molecular defects have been described in the beta globin gene causing beta thalassemia [5-7]. Beta thalassemia is the most common genetic disorder in Pakistan. Thirteen mutations are commonly reported in the Pakistani population, in which the 5 most common mutations include IVS-I-5 (G→C), Codon 8/9 (+G), Codon 41/42 (-TTCT), IVS-I-1 (G→T), and a 619-bp deletion [6, 8]. Thalassemia is a serious disease, requiring a preventive program based on the detection of the beta thalassemia trait (BTT) [8, 9]. Estimation of hemoglobin is important for the diagnosis of certain important clinical conditions. The diagnosis of BTT is based on elevation of Hb-A2 level (>3.5%) [10-12].
Iron deficiency anemia (IDA) is the most common microcytic hypochromic anemia world wide [13, 14]. Iron deficiency modulates the synthesis of Hb-A2, resulting in reduced Hb-A2 levels in patients with IDA [15]. The normal Hb-A2 is composed of 2 alpha and 2 delta chains. The decreased Hb-A2 levels in iron deficiency could be due to decreased transcription and/or translation of the delta gene. Another possible explanation is competition between Hb-A beta chains and Hb-A2 delta chains in binding the limited quantities of available iron to their haem groups, as the ratio of beta: delta chains in normal RBC is 49:1 [16-18].
The present study was designed to determine the role of the co-pathological condition, BTT with IDA in the misdiagnosis of beta thalassemia minor that may lead to childbirth with transfusion-dependent beta thalassemia. This is an important risk factor involved in the propagation of the beta thalassemia gene in Pakistan.
MATERIALS AND METHODS
This multicenter study was conducted at the Baqai Institute of Hematology, Baqai Medical University and the Husaini Institute of Hematology and Oncology Trust, Karachi. Two hundred females aged 16 to 24 years (mean 18.5 years) and having a history of thalassemia in their families, were included in this study.
Venous blood samples were collected into EDTA and basic hematological parameters, hemoglobin electrophoresis, and molecular analysis for beta thalassemia were done on all blood samples. Serum ferritin levels were done only on those samples that were not diagnosed as beta thalassemia minor on cellulose acetate hemoglobin electrophoresis, but were identified at the molecular level, and on samples of patients who were suspected of having IDA during this study.
Basic hematological parameters were performed using an automated analyzer (KX-21; Sysmex, Tokyo, Japan). Cellulose acetate hemoglobin electrophoresis was done using a semi-automated technique (Interlab Roma Microtech Series # 648iso, Rome, Italy).
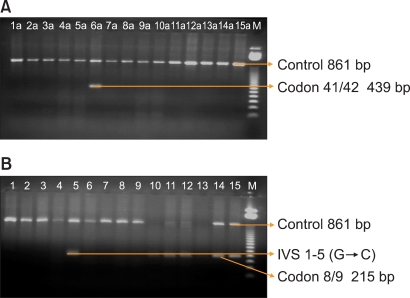
DNA was extracted from whole blood using the Genomic DNA Purification Kit (Gentra system, USA). For the detection of beta thalassemia, the amplification refractory mutation system (ARMS) technique was optimized. Thirteen pairs of primers (normal and mutant) were used for the detection of the beta thalassemia gene that has been previously reported in the Pakistani population [8]. ARMS primers were designed for the detection of both normal and mutant DNA. A control pair of primers was included in each assay. Control primers A, B, and C were amplified at 861-bp fragments from the 3' end of the β globin gene [5, 19, 20].
PCR was conducted with a modified method in a mixture of 10 mmol/L TRIS (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 500 µM each dNTP, 0.2 µM of each primer, 0.5 units of Thermus aquaticus (Taq) polymerase and 0.5 to 1 pg of genomic DNA in a total reaction volume of 20 µL. The modified cycling reaction (DNA thermal cycle; Perkin-Elmer/Cetus) was programmed at 94℃ for 1 minute, 65℃ for 1 minute and 72℃ for 1.5 minutes. After 25 cycles, the samples were incubated for additional 3 minutes at 66℃ [5, 19, 20]. The amplified PCR products were observed using agarose gel electrophoresis, and the mutation was characterized with 100-bp or 50-bp ladder (Fig. 1).
Patients with IDA and patients with the co-pathological condition BTT and IDA (both having low levels of serum ferritin) were treated with oral ferrous sulfate (325 mg/day). These subjects were then followed for a period of 20 weeks. Basic hematological parameters, hemoglobin electrophoresis and serum ferritin were performed on all treated subjects during and at the end of follow-up. Results were analyzed using SPSS statistical software version 17 (Chicago, IL, USA).
RESULTS
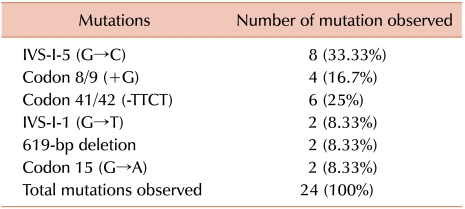
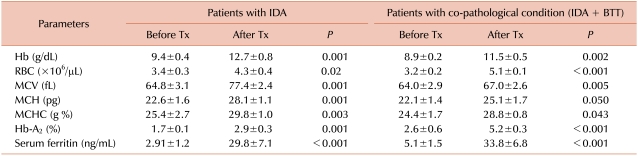
Two hundred unmarried females having a history of thalassemia in their families were included in this study. Of these, 34 were found to be anemic. One of the types of anemia diagnosed was microcytic hypochromic based on red cell indices (MCV, MCH and MCHC). Among the 34 anemic subjects, 16 were diagnosed as beta thalassemia minor with the help of hemoglobin electrophoresis. Molecular analysis, performed in all patients, confirmed this diagnosis but identified 8 additional patients with BTT (Table 1). Eight patients, in which hemoglobin electrophoresis had failed to diagnose BTT, were found with a co-pathological condition, i.e., BTT with IDA, as they have low levels of serum ferritin (Table 2). The 10 microcytic hypochromic anemic patients were diagnosed as IDA based on peripheral blood morphology and low levels of serum ferritin. The patients with IDA also showed low Hb-A2 levels.
The 8 patients with the combined disorder BTT with IDA, who showed normal Hb-A2 levels before treatment, responded to oral iron therapy successfully and showed significant increase in Hb-A2 levels. Patients with IDA only also responded to oral iron and their Hb-A2 levels were found to be normal at the end of the 20th week of iron therapy (Table 2).
DISCUSSION
Beta thalassemia and IDA are the most common microcytic/hypochromic anemias in Pakistan. Pakistan has a population of 160 million people [21, 22]. The annual rate of population growth is 3% and almost 40% of the population is below 15 years of age [23]. There are 5 major ethnic groups Punjabi, Pathan, Sindhi, Baluchi and Urdu [6]. Each ethnic group is subdivided into Castes or 'Biradris' of people ranging from a few thousand to millions [22, 23]. There is a very strong tradition for people to marry within their Biradris. Marriage between close relatives, especially first cousins is a very common custom [22, 23].
Beta thalassemia major is a fatal condition that only occurs when both parents are heterozygous for the beta thalassemic gene (beta thalassemia minor). Therefore, it is important to screen for beta thalassemia minor in the population to stop the propagation of the gene. In Pakistan, almost 6% of the population has the beta thalassemia gene (BTT) [23]. IDA is also the most common anemia in Pakistan, especially in females and young children [24, 25]. Thus, the chance of having a co-pathological condition, i.e., BTT with IDA is very high, and the result of this study revealed a high percentage of such cases.
This study showed the presence of 5 common mutations IVS-1-5 (G→C), codon 8/9 (+G), codon 41/42 (-TTCT), IVS-1-1 (G→T) and 619-bp deletion with a frequency of 91.66% in the Pakistani population. Usman et al. [6] also reported the presence of these 5 mutations with a frequency of 90% in the Pakistani population in a large number of samples (N=400), and proposed it as the basis of a prenatal program on a larger scale to stop the propagation of the beta thalassemia gene. This is an important point that makes the thalassemia prevention program, especially antenatal diagnosis, more affordable at the grass root level in Pakistan, but there is still no reduction in the rate of neonates with transfusion-dependent beta thalassemia. This crucial fact demands re-thinking the factors that play a major role in the propagation of the beta thalassemia gene. Therefore, this study was conducted to identify one of the very important factors that propagate the beta thalassemia gene in Pakistani population. The test population were unmarried females of childbearing age belong to families having a history of thalassemia. The diagnosis of beta thalassemia minor was based on elevated levels of Hb-A2, i.e., >3.5%. This study found 4 cases of BTT that were not diagnosed on Hb-electrophoresis, as they have normal levels of Hb-A2, but molecular analysis identified them as BTT. Further investigation revealed that these patients have the co-pathological condition, BTT with IDA (as they have low levels of serum ferritin). These patients showed significant change in Hb-A2 levels after iron treatment.
This important finding explains that iron deficiency causes a decrease in Hb-A2 levels, as these patients also have beta thalassemia minor, so elevated Hb-A2 levels after reduction became normal levels. This phenomenon is also strongly favored by patients with IDA, as they have low Hb-A2 before treatment, but after treatment, they showed a significant rise in Hb-A2 levels. Thus, it is proved that iron deficiency caused significant lowering of Hb-A2 levels and the patients with the combined disorder BTT with IDA may show normal Hb-A2 levels.
This fact is not negligible, as many factors such as the lack of awareness among working pathologists and clinical consultants, poor facilities for diagnosis and expensive cost of molecular diagnosis (for screening purpose) may result in interpretation of these subjects as normal. These subjects, who may marry a man with BTT, may produce children with beta thalassemia major. This is the most important cause of the propagation of the beta thalassemia gene in the Pakistani population that becomes a serious hindrance for the thalassemia prevention program in Pakistan. To stop the propagation of the beta thalassemia gene through this mechanism, it is critical to identify cases of co-pathological conditions. No case of mild to moderate anemia suspected of having BTT based on hemoglobin electrophoresis will be reported as normal until the probability of iron deficiency is ruled out, especially in a country like Pakistan, where the rate of thalassemia minor is high and the etiology of IDA is common.
 XML Download
XML Download