

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The association between primary immune thrombocytopenia (ITP) and autoimmune hemolytic anemia (AIHA) was first reported by Evans et al. in 1951. This clinical entity is generally referred to as Evans syndrome. Most scholars accept that ITP and AIHA share some pathophysiological mechanisms. Since this first report, there have been many reports of Evans syndrome in association with other autoimmune diseases. Hashimoto's disease is one of the most common autoimmune diseases and is often associated with other non-endocrine autoimmune diseases. Evans syndrome is a non-endocrine autoimmune disease and can present with anti-thyroid auto-antibodies [1]. Although it is assumed that autoimmune thyroiditis and Evans syndrome have overlapping immunological mechanisms, to date, there has been little evidence to support this assumption. Here, we present a 50-year-old female patient who was diagnosed with Hashimoto's disease, AIHA, and ITP over a 16-year period.
The patient was diagnosed with Hashimoto's disease and overt hypothyroidism in May 1994 and AIHA in October 2006. We prescribed synthetic thyroxine and prednisolone for Hashimoto's disease and AIHA, respectively. The prednisolone dose was tapered to 2.5 mg every other day. Subsequently, after 22 months, in September 2009, she developed ITP and was diagnosed with Evans syndrome (the combination of AIHA and ITP).
Treatment options for Evans syndrome include corticosteroids; intravenous immunoglobulin; immunosuppressive drugs such as danazol and rituximab; splenectomy; and stem cell transplantation [2]. Recently, the use of rituximab has shown promising results [2, 3]. Several cases of combined Evans syndrome and thyroid endocrine dysfunction (hyperthyroidism or hypothyroidism) have been reported [1, 4, 5]. However, to the best of our knowledge, this is the first Korean report that describes a case with the sequential development of autoimmune thyroiditis, AIHA, and ITP, and was successfully treated with rituximab.
Go to : 
CASE REPORT
A 51-year-old female patient visited the outpatient clinic of the Department of Hematology for regular follow-up visit for her AIHA in September 2009. She was diagnosed with Hashimoto's thyroiditis and had been taking synthetic thyroid hormone for hypothyroidism since 1994. In 2001, her anti-microsomal antibody level increased to 1:6,400, but we did not have data about her thyroid autoantibody level at diagnosis. In September 2006, she was admitted to the hospital due to dizziness and jaundice, and was subsequently diagnosed with AIHA. Serologic testing showed elevations in anti-microsomal antibody (49 U/mL) and thyroglobulin antibody. Thyroid stimulating hormone (TSH) receptor antibody level was normal (0.3%). The results of a serologic test for anti-nuclear antibody were positive. Both direct and indirect Coombs tests also gave positive results. The tests for human immunodeficiency virus (HIV) antibody, hepatitis B surface (HBs) antigen, HBs antibody, hepatitis C virus (HCV) antibody, and IgM hepatitis A virus (HAV) antibody were all negative.
After this diagnosis, her hemoglobin level waxed and waned between 6 g/dL and 11 g/dL based on the dose of prednisolone being taken at the time. During this time, she refused to undergo a splenectomy.
Abrupt thrombocytopenia (68×109/L) was detected during a regular follow-up on September 1, 2009. During that visit, she also complained of weight gain of 10 kg that had occurred during the past 3 years. On physical examination, she showed no signs of exophthalmos. The abdomen was soft and flat, and the liver and spleen were not palpable. Her platelet count was 179×109/L during her previous visit in July 2009. The patient had persistent thrombocytopenia (21×109/L to 30×109/L) for the subsequent 3 months while she was taking prednisolone (2.5 mg/day). On the other hand, her hemoglobin level remained stable (10 g/dL to 13 g/dL) during the 3-month period. No abnormal findings were observed on bone marrow biopsy, and bone marrow cellularity was normal (40%). Normal levels of erythropoiesis and granulopoiesis were observed, and the number of megakaryocytes was slightly high. There were no cytogenetic abnormalities. On the basis of these results of bone marrow biopsy, she was diagnosed as having ITP. Serologic testing showed elevations in anti-thyroglobulin antibody (>2,000 U/mL) and anti-microsomal antibody (80.3 U/mL), and the serum was positive for the anti-nuclear antibody test (1:640×, cytoplasmic pattern).
After the diagnosis of ITP, the dose of prednisolone was increased to 1 mg/kg (60 mg). While prednisolone was being tapered back down over a period of 4 months, the platelet count decreased to 7×109/L. Rituximab (375 mg/m2) was administered once a week for 4 weeks along with 1 mg/kg prednisolone from May 20, 2010 till June 10, 2010. Rituximab was well tolerated, and the platelet count rapidly increased to 92×109/L by 2 weeks after the completion of the fourth dose of rituximab. Azathioprine and danazol were also added to the treatment regimen. Azathioprine 100 mg was added from June 17 till September 16 (for 3 months) and danazol 300 mg was added from June 29 to August 31. Considering median time to response (usually few months) of these drugs, elevation of platelet count was due to rituximab. Hence, we stopped both azathioprine and danazol. Subsequently, her platelet count was maintained above 100×109/L. Hemoglobin levels, platelet counts, doses of prednisolone, and the timing of medications are illustrated in Fig. 1. Currently, she is not taking any medication for ITP or AIHA and appears to be in complete remission, except for a slightly decreased platelet count (>110×109/L) for about 10 months.
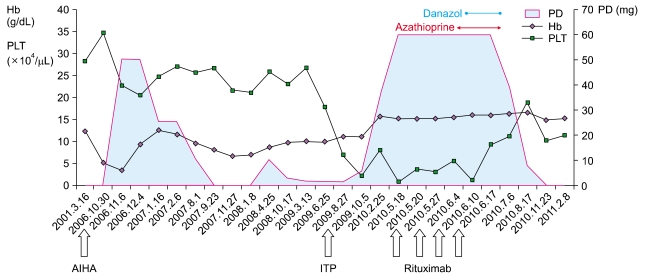 | Fig. 1Trends in Hb level and platelet count with PD dosage and rituximab treatment. PD indicates dose of prednisolone. The units of Hb and PLT in this figure are g/dL and ×104/µL, respectively. Date of diagnosis of diseases and treatment of rituximab is expressed at the bottom of figure.
Increase in dose of PD elevated Hb level but not PLT level. PLT level is increased after the use of rituximab.
|
Interestingly, the intermittent peaking of TSH in this patient was greatly reduced after the use of high-dose prednisolone and rituximab in combination with a low dose of L-thyroxine (0.1 mg). Before the initiation of rituximab, her TSH level fluctuated between 0.17 mIU/L and 19.7 mIU/L, even though she was taking a higher dose of L-thyroxine (0.15 mg). The doses of L-thyroxine, prednisolone, and rituximab, and the TSH levels are presented in Fig. 2. We could not follow up the autoantibody level after rituximab therapy, because the patient was transferred to another hospital. Her last serologic test at our institution showed reduction in the titer of anti-nuclear antibody test (1:80×,cytoplasmic pattern).
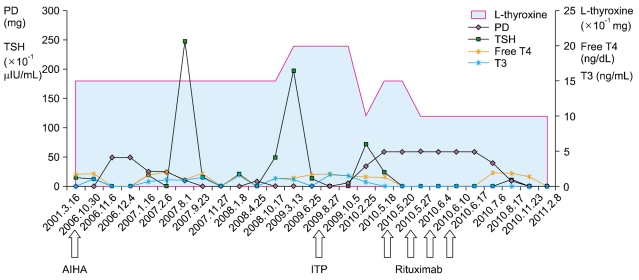
Go to :
DISCUSSION
Evans syndrome is a rare hematological abnormality characterized by Coombs-positive AIHA and ITP without any known underlying cause. This condition may be idiopathic or associated with other diseases. First-line therapy is immunosuppression, while second-line therapy includes danazol and splenectomy [2, 3]. Various immunosuppressive therapies, including Vinca alkaloids, androgens, corticosteroids, intravenous immunoglobulin, and splenectomy, have been used to treat Evans syndrome. However, both medical and surgical treatments have been rather unsuccessful in treating refractory cases of Evans syndrome [6]. Response rates to conventional agents are variable, and some patients require lifelong immunosuppression to keep the disease in remission. Rituximab is a chimeric human/mouse monoclonal antibody that targets CD20 on B lymphocytes [7] and is increasingly used for a variety of autoimmune disorders, including Evans syndrome [2, 3, 8]. Rituximab has also been used to treat disorders associated with autoantibody production [9].
Previously, many researchers have reported the successful use of rituximab for the treatment of patients with corticosteroids-refractory Evans syndrome [3, 7, 10, 11]. There have been several reports of the co-existence of Evans syndrome and thyroid endocrine dysfunction [1, 4, 5]. Recently, there have been reports of the use of rituximab for the treatment of autoimmune thyroid diseases [12-14]. Rituximab induces B cell depletion and decreases the production of thyroid autoantibodies. B cell depletion resulted in diminished B cell and T cell infiltration in the thyroid, thereby inhibiting the development of spontaneous autoimmune thyroiditis. This might serve as a potential mechanism of rituximab in Hashimoto's thyroiditis.
Although we could not determine the thyroid autoantibody level after rituximab therapy, thyroid functions were normalized gradually despite taking a small dosage of L-thyroxine. Our results suggest that rituximab therapy may be beneficial for patients with Hashimoto's thyroiditis. To our knowledge, our report is the first to describe the stabilization of both autoimmune thyroiditis and Evans syndrome by the administration of rituximab. Although the causes of these diseases remain uncertain, this case suggests a common immunological mechanism may play an important role in the pathogenesis of these diseases and supports the use of rituximab in patients with these 3 disorders (Hashimoto's thyroiditis, AIHA, and ITP).
Go to :
 XML Download
XML Download