

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Multiple myeloma (MM) is the most common primary bone cancer, representing 10% of all hematological malignancies and 1-2% of all cancer-related deaths [1], and occurs when malignant of B-cells progressively infiltrate the bone marrow and produce immunoglobulin after clonal expansion [2]. Conventional therapeutic protocols include chemotherapy with bone marrow transplantation and drug treatment involving combinations of melphalan, vincristine, carmustine (bischloroethylnitrosourea), cyclophosphamide, doxorubicin (Adriamycin), thalidomide, and prednisone and dexamethasone [3, 4]. These agents, used as monotherapies or in combination, have significantly improved MM outcomes, but long-term tolerance, graft-versus-host disease, and toxicities associated with some of these drugs represent great limitations [5]. The median survival is still only 3-5 years, and cases of relapse are frequent [6]. New drugs, such as bortezomib (Velcade) and lenalidomide (Revlimid), have recently been introduced as novel and more-curative therapies. However, like with conventional treatments, long-term tolerance and toxicities associated with these drugs are major limitations [5]. Therefore, new therapies are definitely needed.
Historically, remission of hematological malignancies, such as Burkitt's lymphoma and Hodgkin's disease, has been shown to be induced by clinical infection with the measles virus (MV) [7]. This finding paved the way for 2 major therapeutic strategies: The first is based on the use of viruses as oncolytic agents (http://www.hindawi.com/journals/av/2012/186512/), since oncolytic viruses preferentially replicate in tumor cells by taking advantage of cancer-specific cellular changes [8]. This specificity is usually improved by deleting the E1A viral gene that is required for replication [9]. The second strategy, instead, uses different categories of viruses as possible vectors to deliver genes inside human tumor cells. Among the viruses used for viral therapy of tumors, 4 RNA viruses (MV, vesicular stomatitis virus, reovirus, and CVA21 [coxsackievirus A21]) and 2 DNA viruses (adenovirus and VV [vaccinia virus]) have been studied, with the goal of finding a translational application for the treatment of multiple myeloma [10].
The adenovirus vectors have been the most commonly used vectors in human cancer treatment, especially for cancer gene therapy strategies based on intratumoral injection (Supplementary Table 1) [11]. Therefore, the first part of this review focuses on adenovirus and adenovirus-derived vectors and describes the state-of-the-art application of these viruses as oncolytic or gene therapy vectors and the potential therapeutic use of adenovirus vectors for the cure of MM. In the second part of this review, we discuss a significant new cancer antiviral phenotype described by our group that is shared by solid and hematologic malignancies and that may prove to be one of the causes of the partial failure of viral-based therapeutic trials.
ADENOVIRUS AS A VECTOR FOR ONCOLYTIC THERAPY IN MULTIPLE MYELOMA
Adenoviruses are nonenveloped, dsDNA viruses that, in nature, infect cells by binding the fibrous knob of the coxsackie and adenovirus receptor (CAR) expressed on the surface of target cells [11]. As vectors for oncolytic therapies, these viruses have many advantages over other vectors, including the capability of transducing and replicating in dividing as well as non-dividing cells, the ease of manipulation, and a naturally lytic replication cycle, highlighting the usefulness of these viruses for in vitro production and in vivo curative effects (viruses naturally increase the "dosage" while replicating) [12]. As vectors, adenoviruses have a relatively good safety profile and result in a greater survival rate than other therapeutic vectors. Successful treatment of a variety of tumors using adenoviruses has been demonstrated, and they are highly efficient at in vivo gene delivery [11]. Since 1993, more than 300 clinical trials based on adenoviral vectors have been performed [11], with promising outcomes (Supplementary Table 1 and http://www.clinicaltrials.gov/). Although the first clinical results of trials based on adenovirus as an oncolytic therapeutic agent have been promising, showing clinical safety and the feasibility of the approach, studies have also revealed that tumors can acquire a resistance against this type of therapy, and the efficacy of adenoviral treatment still needs to be improved [13]. Since in vitro adenovirus serotype 5 (Ad5) infects cells via the CAR receptor, and CAR [14] and most of the hematologic cells do not express high levels of these receptors, Ad5 was not initially considered as a possible candidate for MM treatment. However, with recombinant technology, adenovirus can be redirected by modification of the viral attachment fiber knob [15], making its use in the treatment of hematological malignancies possible. MM seems to be a better potential target for this therapy than solid tumors since it is easily accessible, with malignant cells found predominantly in the bone marrow and blood [10]. Several serotypes of adenovirus may be suitable as vectors for tumor therapies. Among the different serotypes, Ad5 was the first to be used as a therapeutic alternative for MM in 2007 [16]. In order to investigate the ability of Ad5 to infect myeloma cells, Senac et al. studied the in vitro permissivity of 2 myeloma cell lines, ALMC-1 and ALMC-2, derived from patient samples and representing 2 different stages of disease [17]. Furthermore, by distinguishing tumor cells positive for the expression of the CD138 surface marker from normal bone marrow cells that lack CD138 expression, they confirmed that Ad5 infects a higher ratio of tumor cells in MM. Senac et al. proved that not only Ad5, but also Ad6, Ad26, and Ad48 are capable of infecting and killing the majority of MM cell lines as well as ex vivo primary cells from patients, confirming the feasibility of translating adenovirus-based therapies to MM patients. Their data suggested that adenovirus may retain the ability to selectively kill tumor cells while sparing normal bone marrow cells in the context of MM treatment [18].
ADENOVIRUS AS A VECTOR FOR GENE THERAPY IN MULTIPLE MYELOMA
The second described strategy, taking advantage of the characteristics of adenovirus, is based on the insertion of genes of interest into the genome of a modified adenovirus. In this way, it is possible to use the modified viral particles for delivering genes that are, for example, defective or mutated in the tumor [15], codifying for enzymes that can be then used to activate specific drugs [19], or codifying for proteins able to inhibit the tumor growth directly or indirectly by inducing an immune response [20], specifically to MM tumor cells.
An example of the first strategy (i.e., delivery of defective of mutated genes to the tumor) is found in the work of Torturro, who described adenovirus-mediated cytotoxic gene therapy, showing the efficiency of recombinant adenovirus-p53-mediated cytotoxicity in vitro in Burkitt's lymphoma and MM [15]. In their work, they emphasized the importance of CAR expression and cellular signaling pathways in adenovirus-mediated cytotoxic therapies for MM and other lymphoproliferative malignancies [15]. Using a similar strategy, Ni et al. were able to block proliferation in the MM cell line U266 by expressing the dominant negative inhibitor of κBα (Ad5IκB), blocking in this way the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation [21].
As an example of coding for enzymes that activate specific drugs, Teoh et al. studied the ability of adenoviral vectors to deliver the thymidine kinase (tk) gene into MM cells. This group demonstrated that MM cell lines and MM patient cells express both adenoviral receptors and DF3/MUC1 protein. They hypothesized that the DF3 promoter could be used as a selective promoter to control the expression of therapeutic recombinant genes only in tumor cells [19]. In that study, expression of the tumor-selective promoter DF3/MUC1 was found only in MM-derived cells (MUC-positive) and was absent in hematopoietic progenitor cells (MUC1-negative) [19]. In a combinatory study, the expression of tk in MM potentiated in vivo killing by a combination of viral therapy with ganciclovir [22].
An example of the third strategy is the work of Fernandes et al., who used a conditionally replicating adenovirus containing the CD40 ligand transgene (AdEHCD40L) to demonstrate growth inhibition in MM cells [20]. This strategy was based on previous findings that showed CD40L can directly modulate MM cell growth. Their work has effectively demonstrated that the presence of CD40L is associated with viral oncolysis and results in MM growth inhibition by activating cellular apoptosis [23]. Considering these findings, the clinical application of AdEHCD40L has been proposed in experimental MM treatments [20]. Furthermore, wild type genes for mutated oncogenes can be introduced. For example, Ren et al. designed a vector combining p53 and immunomodulatory molecules, including the GM-CSF (cytokine granulocyte macrophage colony-stimulating factor) and the costimulatory molecule B7-1 (Ad-p53/GM-CSF/B7-1). In 2005, they used this strategy to cotransfer those 3 molecules into MM cell lines and primary myelomas, demonstrating the feasibility and increased immunogenicity of those treated MM cells [24].
OTHER POSSIBLE VIRUSES EXPLORED AS ALTERNATIVE CURES FOR MM
Since the majority of the population has encountered adenovirus at some point in life, and therefore, a rapid humoral immune response versus the virus and the modified agent is generated, the field of viral therapy is also considering other viruses as alternatives for the treatment of MM. Among those, as comprehensively reported by Thirukkumaran and Morris, the VV was first used in 1980 as a virotherapeutic agent in a 67-year-old Japanese patient with IgA MM [25, 26]. Consequently, intravenous injection of the vaccinia strain was found to result in a significant reduction in IgA levels. To date, other clinical trials testing different VV mutants, such as JX-594, have been conducted in patients with metastatic liver cancer. This agent has been considered a possible candidate for clinical trials in hematological malignancies, including MM [27]. Other clinical trials in MM have been performed using vesicular stomatitis virus (VSV) as oncolytic agent. Data from those trials suggests that genetically engineered VSV strains such as VSVΔ51, which has been used in vivo, can be considered as components in potential combination therapies (e.g., with radiation treatment) for MM [28]. Combination therapy with radiation and viral therapy was reported to have good efficacy in a phase I/II clinical trial using reovirus (reolysin) [29-32]. Reovirus was found to be an attractive anticancer therapeutic in further clinical tests for hematological malignancies. Thirukkumaran and Morris anticipated that after encouraging preclinical data is obtained, reovirus will be used in clinical settings in the very near future [10]. Myers et al. reported new ongoing phase I clinical trials with MV for MM. This trial includes a combination of viral and conventional therapies, consisting of intravenous administration of MV-NIS (MV expressing the sodium-iodide symporter) used with or without cyclophosphamide [33]. In this trial, patients are pretreated with cyclophosphamide 2 days prior to MV-NIS injection. Pretherapy and post-therapy hematological and biochemical parameters are yet to be determined, and anti-measles immunity is being monitored. This study also includes serial imagining of virus biodistribution post-123I administration [34]. In unpublished data, Msaouel et al. [35] described encouraging results of this therapy in MM for future phase II/III clinical trials. The MV-Edm (Edmonston-B vaccine MV) strain has also been shown to have oncolytic activity against MM. In experiments employing a transplant model in immune-deficient mice, using different clinical MM samples, this agent successfully killed MM cells [36]. CVA21 is an another potential purging agent, with an ability to selectively target hematological malignant cells [37]. Its specificity is most likely related to the expression of both intracellular adhesion molecule-1 (ICAM-1) and decay accelerating factor (DAF) on the surface of target cells [38, 39]. This virus has already been administered to end-stage MM patients without adverse effects [40], but ongoing human trials are still necessary to evaluate the safety of this therapy.
POSSIBLE OBSTACLES TO VIRAL THERAPIES
Immunogenicity of adenovirus
The injection of adenovirus can lead to the activation of innate and adaptive immune responses against the virus itself. In fact, the strong immunogenicity of this virus is considered one of the major limitations for the in vivo use of this agent. Nayak and Herzog provided a comprehensive overview on the interactions between the immune system and adenoviral vectors [41], suggesting that immune responses against adenovirus can be directed either against the viral protein of the capsid, the vector backbone, or its genomic double-stranded DNA, as demonstrated in gene therapy trials testing the inserted gene.
Systemic delivery of adenovirus vectors results in rapid physiological responses that include activation of innate immunity, induction of cytokines, inflammation, transient liver toxicity, and thrombocytopenia [42]. The innate immune response, through activation of Toll-like receptor (TLR)-2 and TLR-9, stimulates the production of type I interferons (IFNs), resulting in the production of inflammatory cytokines that promote Th1-type immunity with cellular and humoral immune responses [43, 44]. Natural killer (NK) cells are strongly activated by type I IFNs [45] and are known to be mediators of CD4 and CD8 responses. Adenovirus can also induce the innate immune response through MyD88/TLR-dependent and/or MyD88/TLR-independent pathways in different cell types [46, 47]. Part of viral clearance is due to complement opsonization [44] and the generation of inflammation, especially in patients with pre-existing antibodies against adenovirus. Rapid innate activation, as well as the subsequent cytokine storm (IL-6, type I IFNs, RANTES, IL-12 (p40), IL-5, G-CSF, and GM-CSF), stimulate and activate the adaptive immune system [42]. Type I IFN signaling is important for the production of antibodies against adenovirus, and neutralizing antibodies have been found to be effective in blocking innate and adaptive immune responses to the adenovirus.
The generation of humoral immune responses is crucial, since it precludes re-administration of the same serotype. Moreover, more than 97% of humans have pre-existing antibodies against group C adenoviruses as a result of natural infection.
T cells directed against different serotypes have been found in humans. Adenovirus-specific CD4+ T cells recognize conserved epitopes among different serotypes, and it is possible to find these T cells as well pre-activated CD8 cells able to recognize adenoviral epitopes in the circulation of healthy donors. For these reasons, bypassing the immune response to adenovirus seems to be one of the major challenges in the optimization of this novel therapy.
In order to overcome this limitation, several strategies have been utilized, from targeting specific organs, to engineering viral envelopes, switching serotypes, or modifying the transgene cassette. Even immune modulation regimens associated with viral therapy can result in immune avoidance of the viral vector and transgene product, and in some cases, tolerance to the therapeutic gene product can be induced. For example, Mastrangeli et al. showed that the use of subgroup D partially avoided the generation of neutralizing antibodies in a cystic fibrosis trial [48]. Despite the high immunogenicity of adenovirus vectors, which is generally considered a downside in the context of gene therapy, this could possibly prove to be advantageous when developing cancer vaccines since the adenovirus vector may serve as an optimum adjuvant [49].
IFN-STIMULATED GENES, VIRAL STRESS-INDUCED GENES AND THE PHYSIOLOGICAL RESPONSE TO VIRUSES
Viruses physiologically trigger an immediate antiviral innate response that fights viral infection, replication, and spread. In fact, viral pathogens associated molecular patterns (PAMPs) are recognized by TLRs and are activated through IFN regulatory factor (IRF)-3, IRF-5, IRF-7, or NF-κB a transcription factor responsible for the regulation of hundreds of viral stress-inducible genes (VSIGs) that code for proteins with antiviral functions. The TLRs specifically involved in viral recognition are TLR2, TLR3, TLR4, TLR8, and TLR9 [50].
A similar antiviral status can also be induced in uninfected cells, through viral stress-related products originating from neighboring infected cells [51]. In fact, when a virus infects a cell, IFNs are synthesized and secreted as a first line of defense [52]. Transcriptional activation by IFN proteins binding to their specific cell surface receptors leads to the transcription of IFN-stimulated genes (ISGs), whose products inhibit different stages of viral replication [52]. IFN genes encode for a large family of multifunctional, secreted, small regulatory glycoproteins that have important signaling roles in the innate immune response.
There are 3 main types of IFNs: Type I or 'viral' IFNs include IFN-α, IFN-β, IFN-ω, and IFN-τ; type II IFNs include IFN-γ; and type III IFNs, including IFN-λ, are still not well described and have been suggested to be ancestral type I IFNs that also regulate the viral response [53]. Considerable progress has been made in describing the physiological role of IFN signaling components and subsequent antiviral activities [47, 54].
Gene targeting studies have distinguished the 4 main effector pathways of the IFN-mediated antiviral response:
These pathways block viral transcription, degrade viral RNA, inhibit translation, and modify protein function to control each replication step of most viruses [53]. The sets of VSIGs and ISGs that are usually upregulated by viral infection and type I or type II stimulation (Fig. 1) clearly overlap partially [55]. The activation of ISGs promotes the expression of proteins with direct antiviral functions, such as the Mx-resistance-A (MxA) protein that protects infected as well as noninfected bystander cells. MxA proteins are rapidly induced to high levels following IFN or viral exposure and have direct antiviral activity against a wide variety of viruses, including adenovirus [56, 57]. Fig. 1 summarizes the well-studied pathways known to induce upregulation of VSIGs and ISGs.
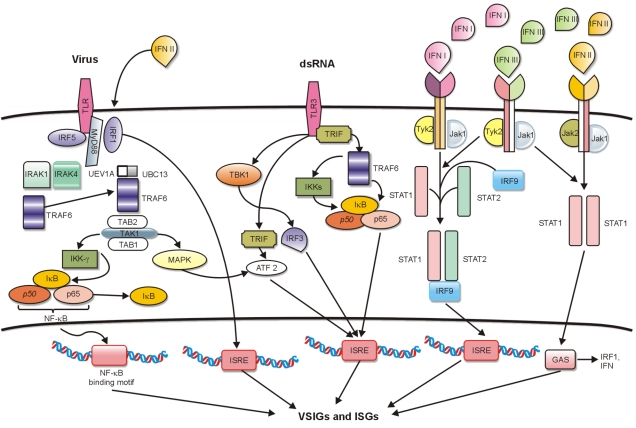
Fig. 1
Different signaling pathways leading to the induction of virus stress-inducible genes (VSIGs) and interferon-stimulated genes (ISGs). Left: virus binding to TLR, stimulating the TLR/MyD88 pathway and leading to the activation and release of NF-κB into nucleus. Center: the dsRNA signaling pathway requires TLR3; there are at least 3 different mechanisms of inducing VSIGs. Right: the JAK/STAT pathway is activated by IFNα, -υ, -β, or -λ; the ISGF3 complex, consisting of STAT1, STAT2, and IRF9, is formed and translocates into nucleus, binding to the ISRE promoter sequence of different ISGs and components of the IFNγ pathway for the activation of GAS promoter sequences. IFNλ mediates the transcription of VSIGs and ISGs through initiation of ISREs or GASs. Abbreviations: TLR, Toll-like receptor; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IFN, interferon; ISGF, interferon stimulated gamma factor; IRF, interferon regulatory factor; ISRE, interferon stimulated response element; STAT, signal transducer and activator of transcription; GAS, interferon-activated site.
![]()
With the molecular characterization of the transcriptional profiles of many tumors, our group and others have reported the existence of 2 subgroups of cancer cells, distinguishable by a spontaneous activation of the ISG molecular profile independent from viral infection or the presence of IFNs in the microenvironment [58-62]. Analysis of this new genomic data has shown that histologically different cancer types, including pancreatic [58], breast, head and neck, prostate, and lung cancer, as well as melanomas and gliomas, generate microarray profiles that identify 2 subgroups distinguishable by specific gene expression of IFNs and inflammatory chemokines [60-63]. In epithelial ovarian cancer, deregulation of JAK/STAT signaling was identified as a cause of discrimination at the molecular level the 2 different subtypes of tumors characterized by the differential expression of ISGs [64]. Several reports have described some ISGs as markers in solid tumors, both in prognostic and diagnostic contexts. For example, in 2006, Andreu et al. showed that IFTM1, one of the most upregulated ISGs following viral infection, was found to be upregulated downstream of β-catenin signal in colorectal tumors [65]. The same marker was found by Gyorffy et al. in ovarian carcinoma, where IFITM1 was actually shown to be associated with therapeutic responses in all treatments analyzed [66]. Weichselbaum et al. proved that, in breast cancer, the VSIG signature is very important for DNA damage resistance and therefore can be used as a predictive marker for chemotherapy and radiation therapy [61].
While ISG overexpression in solid tumors has previously been described, and several reports have shown that the phenotype of the tumor is dependent on this profile, we were the first to associate this phenotype to an in vitro resistance to oncolytic and gene therapy approaches for pancreatic cancer [58] and recently for other types of cancers as well, including MM, ovarian cancer, melanoma, RCC, and colon carcinoma (Raus et al., manuscript in preparation). In fact, we showed that ISG-positive tumor cell lines do not allow either oncolysis or the expression of proteins delivered by adenovirus or adeno-associated virus [58].
In our specific study, we reported for the first time an intrinsic antiviral phenotype in tumor cells that appeared to be independent of the tumor microenvironment, and we performed transcriptional profiling of 3 chronic pancreatitis, 3 primary pancreatic ductal adenocarcinoma (PDAC), 3 paired noncancerous surrounding pancreatic tissues, and 8 PDAC xenografts. We clearly identified 2 distinct PDAC cell phenotypes according to the expression of ISGs, and we found that among several ISGs, 2 phenotypes could be accurately identified by the downstream expression of MxA, which was strongly correlated with the activated antiviral phenotype. MxA, the mediator of one of the first antiviral mechanisms elucidated, is located at a critical intersection of the previously analyzed pathways and is shared by all of these pathways. Therefore, its expression is rapidly induced to high levels when IFN or TLR signals occur [56, 67]. We then expanded our analysis to test the level of MxA expression in 23 human PDACs and 10 human PDAC xenografts by tissue array immunohistochemistry, and we observed constitutive expression of MxA in about 50% of samples. This antiviral state is independent from the tumor microenvironment since it could be confirmed by an in vitro model of human tumors that demonstrated resistance to adenoviral replication and lysis [58]. Furthermore, we found that this status is independent from the presence of IFNs and is caused by spontaneous activation of IFN-stimulated response element (ISRE) sequences [58]. Recently, we published an in vivo study where we confirmed that the antiviral MxA-positive phenotype is acquired by ovarian cancer cells in vivo as an escape to oncolytic adenoviral pressure in a murine model [13]. We believe that the observation of this cancer antiviral phenotype might be of practical significance in stratifying patients as likely or unlikely to respond to viral vector-delivered gene therapy; moreover, it might provide insights into the biology of a subset of human tumors. A thorough understanding of how this antiviral phenotype is generated and if this phenotype may become a new in vivo escape mechanism to viral-based therapies is important in improving the present clinical outcomes of viral therapies.
CONCLUSIONS
Because of the limited efficacy of conventional therapies, new strategies in the treatment of MM are required. Numerous approaches to novel biological therapies are currently under investigation. Remarkable progress has been made in the field of gene therapy, and, thanks to its great potentiality, the techniques of gene transfer are continuously being improved, attracting increasing interest from clinicians. However, many obstacles still need to be overcome, including improvement of transfection efficiencies, targeting to malignant cells, immune system humoral response against the inoculated viral particles, possible tumor escape through upregulation of specific gene sets, and possible toxic viral side effects, which are under evaluation in several completed phase I trials. Human clinical trials using adenovirus in MM patients have not yet been performed; however, determining the safety of these therapies in phase I and phase II trials will be the first step in translating oncolytic adenoviral therapy to MM patients. Although oncolytic virus-based therapies in MM are only beginning to realize their potential, they appear to be promising future treatments for this disease.
 XML Download
XML Download