

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acquired thrombocytopenia, not accompanied by anemia and/or leukopenia (defined as isolated thrombocytopenia), is observed in amegakaryocytic thrombocytopenia [1], infections including human immunodeficiency virus (HIV) infection [2], nutritional deficiencies [3, 4], alcohol ingestion [5], and primary or secondary immune thrombocytopenia (ITP) [6]. A presumptive diagnosis of ITP is made when the history, physical examination, CBC, and examination of the peripheral blood smears do not suggest other causes for thrombocytopenia [7-9]. Bone marrow (BM) examination is not a prerequisite for the diagnosis of ITP in general but may be informative in patients older than 60 years, patients with systemic symptoms or abnormal signs, or patients for whom splenectomy is being considered [6, 7, 9, 10]. Treatment is rarely indicated in ITP patients with platelet counts above 50×109/L in the absence of bleeding, trauma, or surgery [6, 7, 9, 10]. Thus, many patients with isolated thrombocytopenia do not undergo BM examination.
We observed a group of patients with isolated thrombocytopenia, who displayed apparent hypocellularity on BM examination that was not associated with significant dysplasia or megakaryocytic hyperplasia. An efficient diagnosis was challenging in these cases, because hypocellular marrow is not a recognized manifestation of ITP. If these patients had not undergone BM examination, they would have been considered as having ITP. A prospective study on healthy individuals with sustained platelet counts between 100×109/L and 150×109/L (so called borderline thrombocytopenia) revealed that these individuals had 12% probability of developing autoimmune disorders within 10 years [6]. However, the clinical features of isolated thrombocytopenia (platelet counts <100×109/L) accompanied by hypocellular marrow and its natural history have not been studied.
The current diagnostic criteria and recommendations for hematologic disorders originate from Western Europe or North America, where the epidemiology of hematologic disorders is quite different from that of the Oriental countries. For example, aplastic anemia (the most common hematologic disorder showing marrow hypocellularity) is not as prevalent in western countries as it is in the Korean population [9]. The present study was performed to define the clinical features and natural history of isolated thrombocytopenia accompanied by marrow hypocellularity without apparent causes in adults.
Go to : 
MATERIALS AND METHODS
1. Patient eligibility
Adult patients aged 18 years or more with isolated thrombocytopenia, who had hypocellular BM (below 30% in patients aged less than 60 years; below 20% in patients aged 60 years or more) were enrolled at Chungnam National University Hospital between January 2002 and December 2006. Isolated thrombocytopenia was defined as a condition characterized by platelet counts below 100×109/L for longer than 3 months, with normal WBC (more than 4×109/L) and normal hemoglobin levels (12 g/dL or more in women; 13 g/dL or more in men). Pseudothrombocytopenia was excluded by thorough examination of the peripheral blood smear. Only patients with normal karyotype hypocellularity and with no significant dyshematopoiesis (less than 10% in each lineage) or no relative megakaryocytic hyperplasia (less than 5 megakaryocytes on average under a high power field) in the BM study were included. BM specimens were blindly reviewed by 2 independent pathologists. When the cellularities were not concordant, the mean of the 2 different values was accepted as the cellularity. Patients with discordant decisions on dyshematopoiesis or megakaryocyte hyperplasia were excluded. Patients who had viral hepatitis and those who were positive for anti-hepatitis A virus (HAV) IgM antibodies, hepatitis B surface (HBs) antigen, anti-hepatitis C virus (HCV) antibodies, or anti-HIV antibodies were excluded. Patients with autoimmune disorders were also excluded. Patients who were positive for anti-platelet antibody, platelet-associated antibody, antinuclear antibodies, lupus anticoagulants, or anti-cardiolipin antibodies and who displayed low complement (C3 or C4) levels or abnormal thyroid function tests were excluded, and so were patients displaying active infections and pregnant women. Patients with clinical evidence of intravascular hemolysis (reticulocytosis, abnormally high concentration of serum lactate dehydrogenase and indirect bilirubin, and abnormally low concentration of serum haptoglobin) were excluded. Standard single-color flow cytometry was performed for screening of paroxysmal nocturnal hemoglobinuria, and patients displaying more than 3% of CD55- or CD59-deficient RBCs or granulocytes were excluded. In addition to laboratory investigations for determining eligibility, the serum levels of erythropoietin (EPO) were measured by radioimmunoassay at initial presentation.
2. Follow-up
We followed eligible patients who did not undergo platelet transfusion or drug therapy. They were regularly monitored monthly for the first 3 months and then every 3 months for the remaining period. CBC and reticulocyte counts were examined at every visit. Routine chemistry, urine analysis, and studies to rule out systemic lupus erythematosus and primary antiphospholipid antibody syndrome were performed once a year. When the platelet counts decreased further or when pancytopenia developed, BM examination was performed along with conventional cytogenetic studies.
3. Statistical analyses
Statistical analyses were performed using SPSS version 18.0 (SPSS Inc., Chicago, IL). Correlation efficiencies among laboratory parameters were calculated according to Pearson's principle, and P-value<0.05 was deemed statistically significant.
Go to :
RESULTS
1. Patient characteristics
Between January 2002 and December 2006, we studied 224 patients with isolated thrombocytopenia by using techniques including BM examination. Among these patients, 20 (17 men and 3 women) were consistent with isolated thrombocytopenia accompanied by hypocellular marrow. The median age of the patients was 29 years (range, 18-70 years) (Tables 1, 2). Three young male patients aged less than 20 years (UPN 5, 13, and 15) were referred to our hospital for thrombocytopenia found incidentally during routine laboratory examination for military recruitment. The remainder of the group was referred from local clinics for further evaluation of thrombocytopenia found during health examination or during examination for minor illness. One old male patient (UPN 19) had episodic easy bruising, but symptoms or signs commonly associated with thrombocytopenia were not observed in the other patients (data not shown).
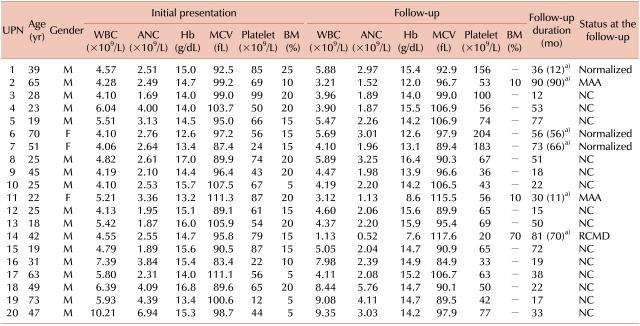
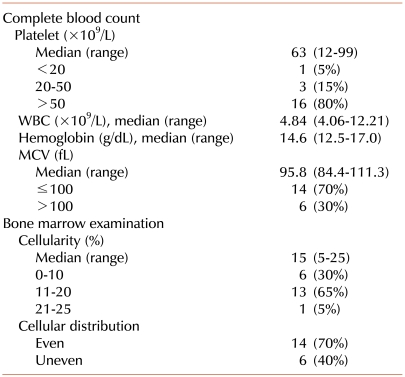
Thrombocytopenia was typically mild, with platelet counts ranging from 12×109/L to 99×109/L (median, 63×109/L). The platelet counts were above 50×109/L in 16 patients (80%) and between 20×109/L and 50×109/L in 3 patients (15%). According to the definition of isolated thrombocytopenia accompanied by hypocellular marrow, anemia was not present in these patients. However, 6 of the 20 patients (30%) had macrocytic red blood cells (mean corpuscular volume, MCV>100 fL). MCV ranged from 83.4 to 111.3 fL (median, 95.8 fL) (Tables 1, 2). Serum EPO levels were measured in 15 patients. Among them, 12 patients (80%) had serum EPO levels higher than the upper limit of the reference range (3.7-31.5 mIU/mL), and 3 patients (20%) displayed levels greater than 3-times the upper limit. No correlation was observed between serum EPO levels and platelet counts (R2=0.0814, P=0.264). In contrast, a weak positive correlation was observed between serum EPO levels and MCV (R2=0.2806, P=0.051) (Fig. 1).
 | Fig. 1Relationship among laboratory parameters in patients with isolated thrombocytopenia accompanied by hypocellular marrow. Weak positive correlations were observed between serum erythropoietin (EPO) levels and mean corpuscular volume (MCV) (B) and between bone marrow (BM) cellularity and platelet counts (C). No correlation was observed between serum EPO levels and platelet counts (A), between BM cellularity and Hb levels (D), or between BM cellularity and serum EPO levels (E).
|
BM cellularity ranged between 5% and 25% (median, 15%) and was 10% or lower in 6 patients (30%), 11-20% in 13 patients (65%), and 21-25% in 1 patient (5%). Cellular distribution was even in 14 patients (70%) and uneven in 6 patients (30%) (Table 2, Fig. 2). There was no correlation between cellular distribution and platelet counts (data not shown). A weak positive correlation existed between BM cellularity and platelet counts (R2=0.2044, P=0.045). However, BM cellularity did not correlate with hemoglobin levels (R2=0.0016, P=0.869) or serum EPO levels (R2=0.0046, P=0.809) (Fig. 1).
 | Fig. 2Representative bone marrow biopsy findings (hematoxylin and eosin [H&E] stain, ×100). (A) Marked hypocellularity with even cellular distribution (from UPN 1). (B) Marked hypocellularity with focally uneven cellular distribution (UPN 15). (C) Moderate hypocellularity with even cellular distribution (from UPN 14).
|
2. Clinical outcomes in long-term follow-up
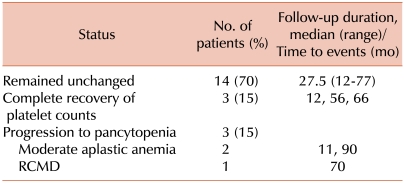
Twenty patients were followed-up for a median of 48 months (range, 12-90 months). Platelet counts in 3 patients (UPN 1, 6, and 7) had recovered to normal values (>150×109/L) during 12, 56, and 66 months of follow-up, respectively. Conversely, 3 patients (UPN 2, 11, and 14) developed pancytopenia at 90, 11, and 70 months of follow-up, respectively. Findings in 2 patients (UPN 2 and 11) were consistent with normal karyotyped moderate aplastic anemia, and 1 patient (UPN 14) was confirmed as having normal karyotyped refractory cytopenia with multilineage dysplasia (RCMD). No severe aplastic anemia, leukemia, paroxysmal nocturnal hemoglobinuria, or autoimmune disorders were observed. The platelet counts remained unchanged in the remainder of patients during the follow-up for a median of 27.5 months (range, 12-77 months) (Tables 1, 3). One patient with aplastic anemia (UPN 11) was placed on immunosuppressive therapy consisting of antithymocyte globulin and cyclosporine, which resulted in complete response. The RCMD patient underwent allogeneic hematopoietic stem cell transplantation and has been healthy with normal CBC for 3 months after transplantation.
Go to :
DISCUSSION
Recently, a significant increase has been observed in incidentally discovered thrombocytopenia, most likely because extensive and regular health examinations are being conducted. Mild to moderate thrombocytopenia is also identified during routine laboratory examination given to young adults during military recruitment. If the relevant causes for thrombocytopenia are not determined in these patients, ITP is the most suitable diagnosis. However, thrombocytopenia is difficult to diagnose, if the BM examination reveals a markedly decreased cellularity without significant dysplasia, suggesting myelodysplastic syndrome (MDS), or megakaryocytic hyperplasia, suggesting ITP. In some cases, the relative increase in the number of megakaryocytes, despite decreased cellularity, is evident. This suggests ITP and a likely response to corticosteroid treatment. In many cases, however, it is difficult to determine the presence of dyshematopoiesis or megakaryocytic hyperplasia because of the paucity of the cells in BM aspirates and marked hypocellularity in BM biopsies.
A decrease in BM cellularity is observed in several hematologic disorders, including aplastic anemia, hypoplastic MDS, and hypoplastic leukemia, and distinguishing one from another is not always easy [11-13]. In the present study, only patients having definite hypocellular marrow, without evidence of megakaryocytic hyperplasia, were included, thereby precluding the possibility that some of the patients might have ITP, because marrow hypocellularity is not a component of ITP. Although screening for Helicobacter pylori (H. pylori) infection is currently recommended in the work-up of adults with typical ITP [9], we did not perform H. pylori test in the present study because the benefits of testing were unproven or controversial during the period of patient enrollment. In future studies, sensitive H. pylori testing will be required.
A prerequisite for aplastic anemia is pancytopenia or bicytopenia, in addition to hypoplastic or aplastic marrow. The patients included in this study did not meet the criteria for aplastic anemia. Patients showing significant dyshematopoiesis or abnormal localization of immature precursors in BM examination were also excluded from the present study, even though dysplasia was difficult to assess in some instances due to the paucity of cells in the aspirates. Immunostain for CD34, CD117, or myeloperoxidase on BM biopsy specimens has recently been reported to be helpful in distinguishing hypocellular MDS from aplastic anemia [13-15]. In addition, high-resolution whole-genome tiling path array comparative genomic hybridization analysis of CD34+ cells from patients with MDS having normal karyotypes can reveal cryptic copy number alterations [16]. However, it remains unknown whether these additional laboratory studies are helpful in further defining isolated thrombocytopenia accompanied by hypocellular marrow. Although we did not perform immunostain or molecular examination, the patients in the present study did not meet the criteria for MDS as proposed by the WHO [17]. Because it is sometimes difficult to diagnose low-risk MDS in patients with mild cytopenia, a provisional diagnosis of idiopathic cytopenia of undetermined significance (ICUS) has been proposed. ICUS was diagnosed in 10 patients with mild to marked unexplained cytopenia without definite signs of a myeloid neoplasm [18]. Since all patients displayed normocellular marrow, the patients in our study could not be classified as having ICUS. Some of the patients showed an uneven cellular distribution in the BM study. The clinical implications of this finding, however, remain unknown, and further studies are required.
In the present study, 2 of 20 patients showed pancytopenia consistent with aplastic anemia during follow-up, indicating a subgroup, in which isolated thrombocytopenia accompanied by hypocellular marrow can be a prodrome of aplastic anemia. These patients tended to display higher levels of serum EPO and larger MCV at initial presentation, suggesting that anemia was not apparent, because erythropoiesis had already been stimulated through a compensatory mechanism. This notion is further supported by a positive correlation between serum EPO levels and MCV. Therefore, a closer follow-up is required in patients who display higher serum EPO levels and larger MCV. One of the 20 patients displayed MDS 7 years later, indicating that isolated thrombocytopenia accompanied by hypocellularity can present as an early manifestation of MDS. Conversely, the platelet counts were normalized during follow-up in 3 patients, suggesting that isolated thrombocytopenia accompanied by hypocellularity is a heterogeneous condition and that a subgroup of this disorder represents a temporary BM depression of unknown cause. These observations suggest that longer follow-up is required even in patients who do not show any changes in CBC during short-term follow-up. If the patients in the present study had not undergone BM examination, they would have been classified as having ITP. Thus, BM examination may be necessary even in isolated thrombocytopenia, at least in the Korean subjects. Taken together, these findings lead us to propose a provisional diagnosis of isolated thrombocytopenia accompanied by hypocellular marrow for patients who display platelet counts below 100×109/L and hypocellularity that is not associated with megakaryocytic hyperplasia or significant dysplasia on BM examination.
In conclusion, isolated thrombocytopenia accompanied by hypocellular marrow encompasses a group of heterogeneous conditions, and its subgroups represent the early manifestations of aplastic anemia, MDS, or temporary BM depression of unknown cause. Regular follow-up is therefore mandatory in patients with this condition, and a large-scale observational study is warranted. In forthcoming studies, vigilant laboratory investigations, including immunostaining for CD34, CD117, or myeloperoxidase of BM biopsy specimens and molecular studies using BM CD34+ cells, may be required.
Go to :
 XML Download
XML Download