

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Polycythemia vera (PV) is a myeloproliferative neoplasm characterized by an absolute erythrocytosis not driven by erythropoietin (Epo), i.e., a primary polycythemia. It is distinguished from other primary polycythemias by the involvement of other hematopoietic cell lineages [1]. Like other myeloproliferative neoplasms, PV is associated with increased risks of evolution to leukemia and of thrombosis [2]; like other polycythemia syndromes, many of its signs and symptoms result from increased blood viscosity [1].
Credit for the first description of PV is generally given to Henri Vaquez, who in 1892 reported a patient with a polycythemia syndrome not due to cardiopulmonary disease [3]. He later proposed that erythrocytosis could be divided into two syndromes: absolute erythrocytosis or polycythemia, resulting from an increased red cell mass; and relative erythrocytosis, resulting from a reduction in plasma volume without increased red cell mass [4]. In 1903, William Osler recognized that several polycythemia cases reported in the literature or encountered in his extensive consulting practice represented a single clinical syndrome not previously delineated, and published a description of this syndrome [5]. The Viennese physician Wilhelm Türk subsequently recognized that PV was not solely as disease of the erythron but involved other hematopoietic lineages [6], and the basic clinical features of PV were largely defined by the time Osler wrote a second, shorter review of PV in 1908 [7]. The history of the early definition of PV has been reviewed in more detail elsewhere [8].
Go to : 
ERA OF CLINICAL/CLINICAL LABORATORY DIAGNOSIS
From its initial descriptions until the last few years, the diagnosis of PV has been made by criteria that were based upon clinical and clinical laboratory findings. In 1966, the U.S. National Cancer Institute funded the Polycythemia Vera Study Group (PVSG) to carry out randomized prospective trials in PV with the goal of defining the best treatment modality [9]. One of the requirements for developing a credible clinical trials program was the establishment of specific criteria for the diagnosis of PV. The subsequent PVSG diagnostic criteria (Table 1) were the basis for all PV diagnostic schema for 40 years [10].

The PVSG criteria essentially reflect the need to establish three features to make the diagnosis of PV. First, the patient must have actual polycythemia and not relative polycythemia (M1). Second, it must be shown that the patient does not have secondary polycythemia (most commonly due to hypoxemia) (M2). Finally, evidence of a myeloproliferative neoplasm needed to be demonstrated (M3 or two minor criteria). The PVSG study criteria are based on clinical or clinical laboratory parameters, other than the requirement for demonstration of an increased red cell mass by nuclear medicine methods. Pathologic parameters (such as bone marrow morphology) were not part of the system, and of course molecular diagnostic parameters were not available in 1966.
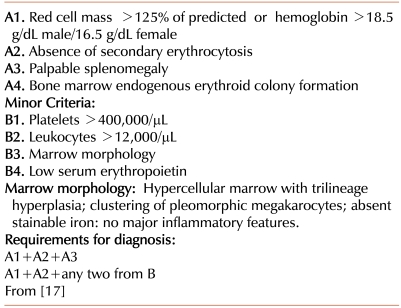
Subsequent developments in research on the pathogenesis of PV have been incorporated into the basic PVSC criteria structure as alternatives or supplements to an original criterion. For example, studies of the coefficient of variation in red cell mass studies has led to the proposal that a red cell mass greater than 125% of predicted value for the local laboratory is a more accurate definition than the specific red cell mass values included in the original criteria [11]. The development of reliable and clinically applicable assays for Epo has led to these assays being suggested as a more conclusive method to rule out secondary polycythemia [12, 13]. Alternative major or minor criteria for evidence of a myeloproliferative neoplasm include splenomegaly only apparent on radiologic studies [11], demonstration of erythroid colony formation in vitro without added Epo (endogenous erythroid colonies, or EEC) [14], increased expression of the platelet receptor Mpl [15], or of gene PRV-1 [16]. However, these alternatives, as expressed in the 2001 World Health Organization (WHO) criteria [17] (Table 2), preserved the basic structure and underlying assumptions of the PVSG criteria.
Go to :
JAK2 V617F IN PV
In 2005, Kralovics and colleagues reported the results of an analysis of the Janus-associated kinase 2 (JAK2) gene on chromosome 9 in 244 patients with myeloproliferative neoplasms (128 with PV) [18]. They observed a dominant gain-of-function mutation in which the valine at position 617 was replaced by phenylalanine (V617F) in approximately half the patients. This mutation appeared to confer an in vitro proliferative advantage upon cells into which it was transfected [18].
Over the next five years, studies of JAK2 V617F experienced proliferation as well. Mutations of JAK2 V617F were reported to occur in 80-96% of cases of PV, and to correlate strongly with the 2001 WHO criteria [19-21]. Studies of PV patients lacking JAK2 V617F reported that a significant number of these had mutations in exon 12 of the JAK2 gene resulting in a myeloproliferative phenotype, although there was some suggestion that exon 12 mutations were more likely to be associated with a picture of pure erythrocytosis [22-26]. Unlike the BCR/ABL mutation in chronic myelogenous leukemia, JAK2 V617F is not pathognomonic for PV but is seen in both essential thrombocytosis (ET) and myelofibrosis [18]. It has been reported that JAK2 V617F expression identifies a subset of ET patients who are predisposed to thrombosis and evolution to myelofibrosis (i.e., more like PV patients) [27, 28]. The allele burden of JAK2 V617F mutations has also been proposed as a tool to distinguish ET (low allele burden) from PV (high allele burden) [29]. The JAK2 V617F allele burden is also associated with response to hydroxyurea, with a high allele burden predicting responsiveness [30], and a reduction in allele burden correlating with clinical response to hydroxyurea [31].
In addition to its role in clinical PV, the identification of JAK2 V617F has had implications for the understanding of the pathogenesis of PV. Transplantation of mice with JAK2 V617F expressing cells leads to a myeloproliferative neoplasm resembling PV [32, 33]. JAK2 V617F appears to be required for the characteristic Epo hypersensitivity of PV erythroid progenitors [34]. However, acquisition of JAK2 mutations does not appear to be the initial molecular event in the pathogenesis of PV, but is rather downstream from some as yet undetermined initiating event [35, 36].
Go to :
JAK2 V617F IN PV THERAPY
As noted above, JAK2 mutation status can serve as a predictor of response to treatment with hydroxyurea [30], and changes in JAK2 V617F status are associated with response to either hydroxyurea or interferon [31, 37]. Reports of agents targeted to JAK2 in PV are largely limited to studies using various PV model systems [38-40] or to clinical trials in post-PV myelofibrosis [41, 42]. The variable results found in the reported clinical studies may be either agent-specific or specific to the patient subset selected for the study.
Go to :
JAK2 V617F AND THE ERA OF PATHOLOGIC/MOLECULAR DIAGNOSIS
As discussed earlier, JAK2 V617F is not pathognomonic of PV but rather is a hallmark of BCR/ABL-negative myeloproliferative neoplasms. Therefore, if it was to be incorporated in a diagnostic schematic analogous to the PVSG criteria, it would be an alternative to M3, which indicates a myeloproliferative neoplasm.
However, the 2008 modification of the WHO diagnostic criteria for PV [43] (Table 3) departed substantially from the underlying concepts of the PVSG criteria. The first major criterion (A1) remains the demonstration of an increased red cell mass, but employs a widely, though not universally [44], accepted criterion based on hemoglobin concentration. The diagnosis of PV is then made by the finding of two additional criteria. This requirement can be met by demonstrating any two of the following: a low serum Epo concentration, a characteristic bone marrow morphology, the presence of EEC, or JAK2 V617F (or a functionally similar mutation, like JAK2 exon 12). This departs substantially from a reliance on readily available clinical and clinical laboratory parameters to more complex testing. PV has become less a clinical diagnosis and more a pathologic and molecular diagnosis.

Go to :
HOW IS JAK2 V617F USED IN ACTUAL PRACTICE?
JAK2 testing has been suggested as the initial step in the evaluation of erythrocytosis [45]; however, there is no study describing how the general population of hematologists uses this test which could be compared to the report by Strieff et al. in 2002, which describes the diagnostic and management practices prevalent in the early post-PVSG era [46]. In a recent report, the use of JAK2 V617F in a single academic practice was described [47]. Patients newly presenting with a diagnosis of PV were routinely tested for the mutation. Of established patients, patients who did not meet the 2001 WHO diagnostic criteria fully tended to be more likely to be tested for JAK2 V617F mutation; patients who seemed to have a clearly established diagnosis.
Go to :
CONCLUSION
The discovery of the JAK2 V617F mutation, the definition of its frequency in PV and other myeloproliferative neoplasms, and the identification of its pathophysiologic implications, have transformed the diagnostic approach to PV from one based in the clinic and clinical laboratory to a truly molecular paradigm. JAK2 V617F has the potential to guide therapy of PV and other BCR/ABL-negative myeloproliferative neoplasms, whether as a therapeutic target or as a marker of response to therapy or of progression risk.
Go to :
 XML Download
XML Download