

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Langerhans cell histiocytosis (LCH) has a variable clinical spectrum, ranging from isolated bone or skin lesions to life-threatening multisystem involvement [1]. Despite the markedly poorer prognosis of patients with multisystem disease and the involvement of risk organs, there are no clear morphological differences among lesions observed in the different clinical categories [2]. LCH has a wide spectrum of clinical features, although the cause of the disease is obscure [1-3]. In order to treat LCH appropriately, it is essential to investigate the disturbances in cell proliferation and apoptotic pathways that may lead to LCH, and to identify the molecules associated with multisystem disease and disease progression [3, 4].
The clonality of cell populations observed in LCH suggests a genetic basis for this disease [2, 4-6]. Genetic alterations at the cellular level may disrupt mechanisms controlling the proliferation and apoptosis of Langerhans cells (LCs). Previous studies have examined the expression and functional significance of LC-specific genes [2, 4-6]. However, only a few studies have examined the genes involved in the cell cycle of LCH cells, such as p53, MDM2, p16, p21, ki-67, and Bcl-2 [4-6]. Thus, much remains unclear regarding the expression of these genes and their clinical significance in LCH.
A series of structurally related enzymes regulate progression of the cell cycle from the G1 to S phase: cyclin regulates the activation of cyclin-dependent kinases (CDKs); CDKs regulate retinoblastoma protein (pRb) and induce the release of E2F transcription factors and the expression of genes required for the S phase; and cyclin-CDK complexes are negatively regulated by a family of kinase inhibitors [7, 8]. The p16INK4 (CDKN2A) gene located on chromosome 9p21 encodes the p16 protein, which inhibits CDKs and blocks cell cycle progression [7-9]. Notably, p16 is often mutated or inactivated in primary tumors, including leukemias, lymphomas, gliomas, lung carcinomas, colon cancer and many cancer cell lines, and is thus regarded as a tumor suppressor gene [10-13].
This report describes the clinicopathologic features of LCH with regard to the p16 expression of LCs in biopsy specimens, which we used to determine whether p16 expression is a relevant clinical risk factor in LCH patients.
MATERIALS AND METHODS
1. Patients
Formalin-fixed, paraffin-embedded biopsy specimens from children with LCH diagnosed between March 1987 and February 2008 at the Asan Medical Center and Chungnam National University Hospital were examined using p16 immunohistochemistry. We analyzed the relationship between p16 protein expression and clinical features. Single-system involvement was defined as uni- or multifocal involvement of a single organ system, whereas multisystem involvement was defined as the involvement of multiple organ systems, with or without organ dysfunction [1-3]. Risk organs were the liver, spleen, lung, and the hematopoietic system [1-3]. Patient medical records were reviewed retrospectively for organ involvement at diagnosis, disease course, relapse, and late sequelae.
2. Immunohistochemistry
Immunohistochemistry was performed on specimens from all cases of LCH using standard laboratory methods. Briefly, 5-mm sections were cut from representative tissue blocks and mounted on silane-coated slides, deparaffinized in xylene, and then rehydrated through p16 (clone JC8; Neo Markers, Fremont, CA). Immunohistochemical staining was evaluated by an experienced pathologist blinded to the clinical outcome.
3. Grading
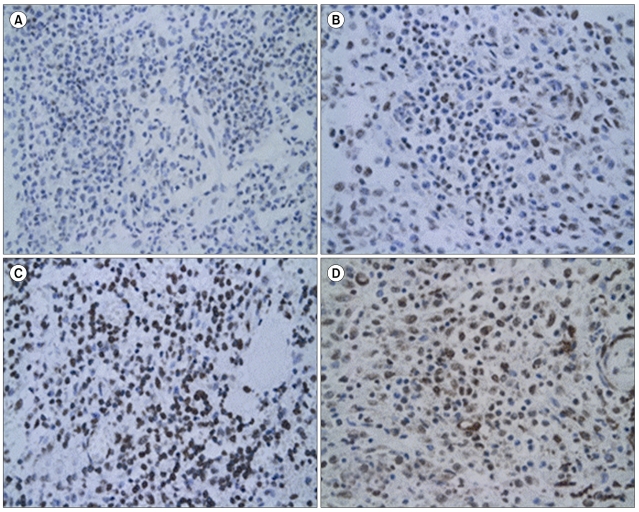
Grading was performed in fields in which LCs were present in compact sheets. LCs were identified morphologically and immunohistochemically by comparing the section stained with the relevant antibody with adjacent hematoxylin and eosin (H&E)- and CD1a-stained sections. Staining was considered positive when it was cytoplasmic or nuclear. At least 500 LCs were counted on each slide using a grid eyepiece at ×400 magnification. The numbers of LCs expressing the relevant antigen were then counted in the same fields and the result was expressed as the degree of staining in LCs compared to that in lymphocytes surrounding the LCs. A semi-quantitative evaluation was made using the following grading system: negative, no staining; ±, weakly positive; 1+, staining similar to lymphocytes surrounding the LCs; 2+, stronger staining than lymphocytes; 3+, much stronger staining than lymphocytes (Fig. 1). For cases with biopsies from more than one lesion, a mean grade was calculated.
4. Statistical analysis
All statistical analyses were performed using SPSS 13.0 (SPSS, Chicago, IL). We attempted to fit an exact multivariate model to the data, but the sample size was too small to achieve convergence. We were cautious about interpreting non-significant results because of the low statistical power of a small sample size. The parameters of the patients at diagnosis were compared between groups using the Mann-Whitney exact test. The event-free survival rate was estimated using the Kaplan-Meier method. An event was defined as the occurrence of relapse, progression, or death. The difference in survival rates between groups according to p16 expression status was compared using the log-rank test. P-value<0.05 was regarded as statistically significant.
RESULTS
1. Patient characteristics
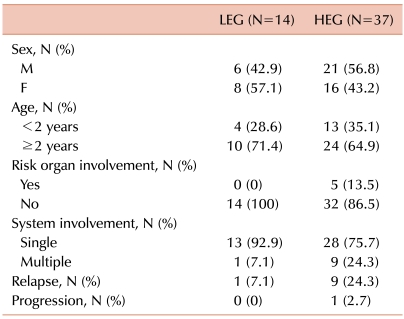
Slides from 70 patients (32 girls, 38 boys) were available for immunohistochemistry, but only 51 patients (24 girls, 27 boys) were included in this study. The data from 19 patients were not suitable for evaluation because the quality of staining was poor, the tumor cells were too difficult to differentiate, or no data at diagnosis were available. The median age of the 51 patients was 49 (range, 0.6-178) months. The diagnosis of LCH was made by demonstrating pathological LCs and a characteristic infiltrate. LCs were identified by a positive immunohistochemical reaction to S100 and CD1a antibodies. The organs involved included bone, skin, liver, lung, bone marrow, pituitary gland, and lymph nodes. Bone involvement with or without the involvement of another site was the most common manifestation and was observed in 90.2% of the cases (46/51). Forty-one of the patients (80.4%) were diagnosed with single-system disease and 10 patients (19.6%) with multisystem disease. Five patients (9.8%) had risk organ involvement at the time of diagnosis.
2. p16 immunohistochemistry
All but 2 cases (96.1%) showed diffuse nuclear and cytoplasmic staining of pathologic LCs. Multinucleate giant cells also showed variable reactivity. According to our grading system, 2 patients were negative, 12 were ±, 14 were 1+, 13 were 2+, and 10 were 3+. To determine whether the level of p16 expression can be used to predict the outcome of LCH, the immunohistochemical expression of p16 was divided into lower expression (negative and ±) and higher expression (1+, 2+, and 3+) groups (LEG and HEG, respectively).
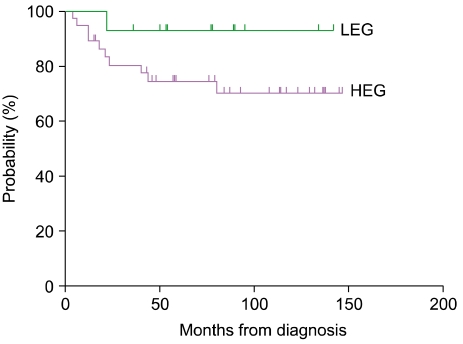
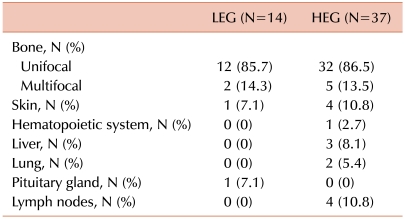
Fourteen of the 51 patients (27.5%) fell within the LEG and 37 patients fell within the HEG (72.5%). All 5 patients with risk organ involvement were included in the HEG. Patients in the HEG also showed a greater tendency of multisystem involvement and relapse, although the difference was not significant (Table 1). Bone involvement was the most common manifestation in both groups, and there were 2 (14.3%) and 5 (13.5%) patients with multifocal involvement in the LEG and HEG, respectively. Although the LEG showed involvement of the skin and pituitary gland (low-risk organs), the HEG showed primarily involvement of the liver, bone marrow, lung, and lymph nodes, all of which, except the lymph nodes, are risk organs (Table 2). The median follow-up duration was 79 months (range, 15-147). The probability of 6-year EFS for the LEG patients was 92.9±6.9%, whereas that for the HEG patients was 70.3±8.1%; however, the difference was not statistically significant according to the log-rank test (Fig. 2).
DISCUSSION
The clinical manifestations of LCH vary considerably and can involve nearly every organ of the body [1-3]. Conventional histology alone can give only a presumptive diagnosis of LCH. The course of the disease is unpredictable, varying from spontaneous regression and resolution to rapid progression and death or recurrence [1-3]. Patients with localized disease generally have a good prognosis and need minimal or even no treatment. In contrast, multisystem disease, which is particularly frequent in children younger than 2 years, carries a risk of poor outcome [1, 3, 14]. In one large study of 101 children with LCH, the overall survival rate was 79%, 74%, and 71% at 1, 3, and 5 years, respectively. Conversely, in patients with liver or spleen involvement, the 1-year survival was 33% and the 5-year survival was just 25% [15]. Therefore, factors influencing the outcome of LCH must be identified to prevent or facilitate the treatment of disease progression and relapse.
The detailed pathogenetic mechanism of LCH remains unknown. Furthermore, it is not yet understood why some patients develop single-system disease, whereas others develop fatal multisystem lesions. The finding that essentially all cases of LCH exhibit clonal LCs suggests a primary genetic alteration [2, 4, 16]. There is much debate as to whether the observation of clonality in lesional LCs proves that LCH is a 'neoplastic' disorder, because clonality is one of the key features characterizing nearly all cancers. Nevertheless, clonality alone is insufficient to make a diagnosis of cancer, as there are examples of clonal 'reactive' lesions [3, 17].
Several studies have suggested that genetic mechanisms play a role in LCH pathogenesis [2, 4-6]. This possibility was further supported by Chikwava et al. [18], who analyzed fractional allelic loss (FAL) across a number of tumor suppressor genes, comparing FAL at various clinical stages of LCH and in LCH cases involving organs at various degrees. They found that the mean FAL in multisystem LCH was significantly higher than that in single-system LCH. Furthermore, FAL was also significantly higher in children with high-risk or special-site LCH than in children with low-risk LCH. These findings suggest that genetic alterations, specifically an increased frequency of loss of heterozygosity (LOH) in high-stage, high-risk forms of the disease, lead to disease progression, potentially due to alterations in tumor suppressor genes also involved in tumorigenesis [18, 19].
Because a genetic component is suspected to play a role in the pathogenesis of LCH, several groups have investigated LC-specific genes and their functional significance in LCH [2, 4-6]. However, few studies have examined genes involved in the cell cycle of LCH cells. The presence of clonal cell populations, as observed in most LCH lesions, may result from a genetic alteration that affects cellular mechanisms controlling proliferation and apoptosis [2, 4, 18]. Several genetic markers of LCH have been identified in previous studies, but their prognostic significance remains unclear.
The p16-Rb pathway is important for detecting DNA damage and inducing cell cycle arrest [7, 9, 10]. This pathway was previously found to be active in LCH lesions [5]. The p16 protein is an important tumor suppressor that is inactivated in a large proportion (>30%) of human tumors [7-11]. This tumor suppressor gene inhibits the CDK4-6 kinases, thus preventing CDK-mediated phosphorylation of Rb and, subsequently, cell cycle progression [20]. The tumor suppressor protein Rb, which associates with several transcription factors in its unphosphorylated or hypophosphorylated form, silences their transactivation functions. The most important transcription factors that Rb silences are the E2F proteins, which activate the expression of important cell cycle proteins, such as cyclins E and A. When Rb is phosphorylated, it will no longer bind E2F, leading to the activation of DNA synthesis [7, 8, 20]. In our study, p16 was expressed diffusely in all but 1 specimen, and patients with specimens showing overexpression had a greater likelihood of poorer prognosis.
Bcl-2 is considered to be a negative regulator of apoptosis, as it inhibits cell death by evoking cell survival signals [21, 22]. Schouten et al. [5] reported that 29 of the 30 LCH cases were positive for Bcl-2, ranging from limited focal reactivity in some cases to strong diffuse staining in others. Savell et al. [23] also observed Bcl-2 protein expression in 11 of 13 cases of LCH; notably, 5 cases showed a germline configuration of the Bcl-2 gene. Furthermore, Amir et al. [2] have founded that Bcl-2 is highly expressed in patients with multisystem disease compared with single-system disease. The overexpression of Bcl-2 may play a role in the activation of p53 and p16 and the subsequent arrest of apoptosis [24-26]. However, it is unclear why Bcl-2 expression is so extensive in LCH lesions, and other markers associated with p16 overexpression should be studied to clarify the pathogenesis of LCH. In immunohistochemical experiments such as ours, it is difficult to assess the coexpression of markers within such a heterogeneous cell population. Consequently, the interpretation of these findings requires a cautionary approach.
This study of the expression of p16 in LCH aimed to define genetic and functional characteristics of LCH that may account for the pathogenesis of the disease. We observed diffuse expression of the p16 protein in both single- and multisystem lesions in patients with LCH. A greater tendency to relapse was observed in the HEG than in the LEG. The HEG had a greater tendency toward multisystem disease and risk organ involvement. However, the association between p16 expression and patient age was not significant. Due to the relatively low incidence of risk organ involvement or relapse in this study cohort, we were unable to detect a clear statistical difference regarding these parameters between the LEG and HEG. Nevertheless, p16 expression may be involved in the pathogenesis of LCH and have predictive power regarding clinical outcome and prognosis. However, additional immunohistochemical analyses examining the functional relevance of this protein and a number of other genetic markers are required to prove this hypothesis. More detailed information regarding genetic markers will help to clearly delineate the different entities of LCH, and may facilitate the development of an advanced classification system as a basis for selecting an appropriate therapeutic approach.
 XML Download
XML Download