

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The survival of acquired aplastic anemia (AA) patients has shown a marked improvement due to immunosuppressive therapy (IST) and bone marrow transplantation (BMT). This improved survival creates the opportunity for observing the long-term course of bone marrow failure syndrome [1, 2]. Technological advances in methods for obtaining complete chromosomal preparations during metaphase and identifying cytogenetics (e.g., various banding techniques, fluorescent in situ hybridization) have allowed cytogenetic studies to be included in the diagnostic workup and have contributed prognostic value in several hematological disorders [3].
Cytogenetic abnormalities (CAs) can be observed at initial diagnosis of AA, may develop spontaneously, or may develop after years of IST [4-6]. Genomic instability does not appear to be a rare event in AA, since transient or permanent chromosomal aberrations have been reported with various onset times. Hence, numerous attempts have been made to determine the clinical implications of CA in AA related to disease prognosis and to possible evolution into clonal hematologic disorders [7-13]. Some researchers contend that since AA evolves into acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) in approximately 3-13% of all cases [7], the clonal cytogenetic events in AA over the course of the disease, as well as at diagnosis, are strongly suggestive of its preleukemic nature [14-17]. Young patients with hypoplastic MDS, especially those with normal cytogenetics, responded well to IST, which suggests that AA and MDS may share the same immune pathophysiology for the development of pancytopenia [18]. Others insist that the CAs in AA are associated neither with the natural course nor with the premalignant characteristics of the disease [8].
The paucicellularity of the bone marrow makes differential diagnosis among hypoplastic MDS [19], hypoplastic AML [20], and AA difficult. Some insist that typical CA, regardless of bone marrow morphology, could discriminate hypoplastic MDS from AA [21], but others consider hypoplastic bone marrow with discrete dysplastic features as hypoplastic MDS [22]. The discrimination between AA and preleukemia is important, because they have different natural courses and warrant different treatment strategies [23]. Therefore, our aim was to determine the significance of CA in adult acquired AA by investigating patient survival and progression to other hematologic diseases.
Go to : 
MATERIALS AND METHODS
We retrospectively observed 222 adult patients with acquired AA, who were treated in 4 hospitals located in Busan, South Korea, between January 1995 and March 2009. Patient information was obtained by reviewing medical records, according to the protocols approved by the Institutional Review Board (IRB number: 09-10-90). The results of cytogenetic studies were available for 152 patients.
Chromosomal analysis was performed on 24-48 h synchronized cultures of bone marrow cells. Karyotyping was done using standard Trypsin-Giemsa banding methods. Diagnosis and assessment of the disease were based on the modified Camitta's criteria. Severe AA required at least 2 of the following criteria: less than 0.5×109 neutrophils/L, less than 20×109 platelets/L, or less than 1% reticulocytes (corrected for hematocrit) in the peripheral blood; marked hypocellularity (less than 25% of normal cellularity) or moderate hypocellularity (25-50% of normal cellularity), with less than 30% hematopoietic cells remaining in the bone marrow aspirate [24]. Non-severe AA was defined by hypocellular bone marrow with pancytopenia (without other criteria of severe AA). Morphologically hypoplastic bone marrow with dysplastic cells was considered as hypoplastic MDS and excluded. Clonal CA were defined as 2 or more cells showing the same chromosomal gain or structural abnormality, or 3 or more cells with the same chromosomal loss. At follow-up, the presence of only 1 abnormal cell was considered sufficient to demonstrate clonal abnormality [25]. A cytogenetic study was considered to have failed, if fewer than 5 metaphases were seen despite inspection of at least 4 slides. Of the 152 patients with available cytogenetic studies, 25 patients (15%) were excluded from the analysis because of a lack of meaningful results (e.g., culture failure and insufficient number of metaphases).
We grouped patients according to the CA detected at initial diagnosis or at follow-up and according to progression to other hematologic disorders. Group 1 included patients with a normal karyotype at diagnosis and during follow-up, without evolution into clonal hematologic disorders. Patients in Group 2 demonstrated a normal cytogenetic profile at initial diagnosis, but acquired clonal evolution later, and Group 3 included patients with CA at initial diagnosis, regardless of follow-up cytogenetics.
Means and medians were calculated when appropriate, continuous variables were compared by the student t-test, and categorical variables were analyzed by the chi-square test. The cumulative survival was estimated by the Kaplan-Meier method. The survival rates of patients with normal and abnormal cytogenetics were compared by the log-rank test.
Go to :
RESULTS
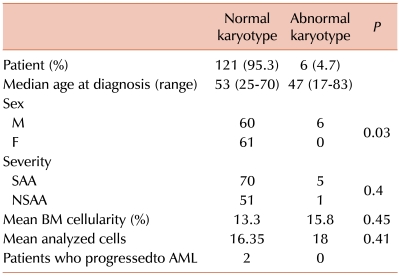
We analyzed data from 127 patients on whose samples successful culture had been performed. The median follow-up period was 46.8 months (range, 0.3-165.8 months). One hundred and twenty-one patients (95.3%) had normal karyotypes and 6 (4.7%) showed abnormal karyotypes at the first chromosomal analysis. Other than sex, there were no significant differences in baseline characteristics between the normal and abnormal karyotype groups. The median age of the latter group was 53 years (range, 25-70), compared to 47 years (range, 17-83) for those patients with a normal karyotype. All 6 patients with abnormal karyotypes were male, and the male to female ratio was 1:1 in the remaining patients. The ratio of severe AA (SAA) to non-severe AA (NSAA) for patients with abnormal karyotypes at initial diagnosis was 5:1, as compared to 1.4:1 in the remaining patients. The mean bone marrow cellularity was 13.3% vs. 15.8%, and the number of mean-analyzed cells was 16.4 vs. 18.0 for the normal and abnormal karyotype groups, respectively.
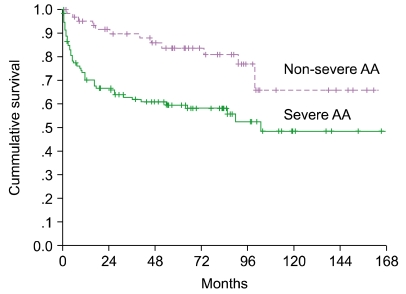
Patients with abnormal karyotypes at diagnosis showed no evolution to hematologic disorders; however, of those with normal cytogenetics at diagnosis, 2 progressed to AML with clonal evolution (Table 1). The median survival according to the severity of AA was 103.3 months in SAA vs. not yet reached in NSAA (P=0.0014) (Fig. 1).
Among the patients with normal cytogenetics at initial diagnosis, 117 patients exhibited normal cytogenetics, while continuing to show AA during follow-up (Group 1). Patients in Group 1 were treated with IST or BMT in case of SAA.
The 4 patients in Group 2 demonstrated normal cytogenetics at initial diagnosis but developed CA during the follow-up period. Of these, 2 did not progress to AML or MDS at the time of clonal evolution. They showed monosomy 4 and monosomy 7 after 126.0 and 17.7 months of supportive care, respectively. Both were females (25 and 68 years old) at diagnosis, respectively. At initial diagnosis, the patient with monosomy 4 was found to have NSAA, and the patient with monosomy 7, SAA. They are still alive, having survival times of 151.0 and 46.2 months, respectively. They do not require transfusions, nor have they developed any other hematologic malignancies. The other 2 patients progressed to AML with structural CA during follow-up assessment. One of these patients showed complex CA after 2.6 months of supportive care, and the other patient was characterized by t(1;11), 51.7 months after IST. They received chemotherapy for AML, but both died of sepsis 3 months after AML diagnosis. The median intervals between the secondary clonal hematologic disorders and death were 2.6 and 2.9 months, respectively, and their survival time was 5.2 and 54.6 months, respectively.
The 6 patients in Group 3 were diagnosed with AA, and all demonstrated CA at initial diagnosis. CA was found to be +8 in 2 cases and +11 in 1 case, -Y in 1 case, t(2;9)/t(1;20) in 1 case, and t(22;?) in the remaining case. The patient with +11 received IST and showed a CA of +1 after 23 months. He is still alive and does not require transfusions. The patient with t(2;9)/t(1;20) and 1 of the patients with +8 also received IST, but died due to sepsis after 16.4 and 16.8 months, respectively. The patient who had -Y was treated with granulocyte colony-stimulating factor (G-CSF) only, and the patient died of sepsis after 3.3 months. The remaining patient with +8 and the patient with t(22;?) received intermittent transfusions and supportive care, and both are still alive. None of the patients in Group 3 progressed to AML or MDS (Table 2). Patients in Group 1 did not reach the median survival time; their mortality rates were 0.19 at 1 year, 0.27 at 2 years, and 0.53 at 5 years. Patients in Group 3 showed a median survival of 16.8 months (P=0.37, compared with Group 1).
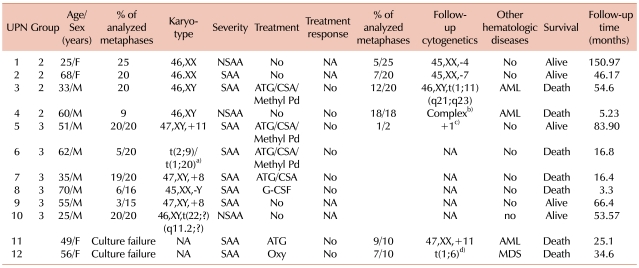
Table 2
Characteristics of patients with cytogenetic abnormalities or progression to hematologic malignancies.
a)46,XY,t(2;9)(q33;p24)[3]/46,XY,t(1;20)(p10;q10)[2], b)46,XY, t(6;7)(q10;q10),?t(11;18)(q13;q21),der(21)t(21;22)(q22;q11.2),-22,+2 mar, c)46,XY,?+1,?der(1;7)(q10;p10)[1], d)46,XX,der(6)t(1;6)(q12;p25).
Abbreviations: NA, not-available; ATG, anti thymocyte globlulin; CSA, cyclosporine; Methyl Pd, methylprednisolone; G-GCF, granulocyte colony-stimulating factor; Oxy, oxymetholone; Other hematologic diseases, evolution into other hematologic diseases; AML, acute myeloid leukemia; MDS, myelodysplastic syndrome.
![]()
Two patients, who were not included in the analysis because of an unsuccessful culture at initial diagnosis, progressed to AML or MDS. One of the patients progressed to AML with +11 after 12.8 months of IST and died of sepsis 12.2 months after AML diagnosis. The other patient progressed to MDS with t(1;6) after 28.3 months of oxymetholone treatment but died of chronic GVHD 6.3 months after BMT.
Go to :
DISCUSSION
Previous studies have estimated CA and genomic instability of AA (at diagnosis or at follow-up) in approximately 4-15% of all cases, but the clinical significance of CA in AA is still unknown [4-6]. In our study, a review of bone marrow pathology and of cytogenetics in patients typically presenting AA by performing peripheral cytopenia and marrow pathology showed that the majority of patients had normal cytogenetics (Group 1), and only 6 patients presented with clonal CA at initial diagnosis (4.7%) (Group 3). Although the number of patients analyzed in this study was too small and the observation period too short to generalize, the clinical characteristics and the survival data trends demonstrate a male predominance, an increasing severity, and a tendency of shorter survival in Group 3 patients than in Group 1 patients.
CAs (e.g., +8, -Y, +11, other structural abnormalities) are commonly associated with AML or MDS [26, 27]. These CAs were also observed concurrent with AA diagnosis in our study. Although only 1 patient in Group 3 underwent repeated bone marrow examinations, a review of repeated peripheral blood smear revealed that none of the patients progressed to other hematologic malignancies. Two patients in Group 2 later developed numerical CA (monosomy 4 and monosomy 7), but they did not develop AML or MDS. Although the number of patients in Groups 2 and 3 is too small to form conclusions, the clonal cytogenetic events in AA patients does not necessarily imply that AA is a preleukemic syndrome, nor that CA presenting with AA predicts subsequent AML or MDS development (regardless of IST). In addition, the full leukemogenic implications of +8 and -Y are undetermined; however, sole +8 is an insufficient condition for leukemogenesis [28], and the loss of the Y chromosome has been observed in the general elderly population, with an incidence similar to that reported for AML or MDS [29]. The true leukemogenic potential of these chromosomes needs to be investigated. The 2 patients in Group 2, who developed monosomy 7 and monosomy 4 during follow-up, showed long-term survival in our study. Although the number of patients is too small and the lack of follow-up cytogenetics in patients with initial CA makes it difficult to generalize, the acquisition of monosomal karyotypes in otherwise typical AA patients may not necessarily mean a poorer prognosis.
CAs may be adopted by some experts to discriminate between AA, hypocelluar AML, and hypocellular MDS [30]. Hypocellular bone marrow may obscure the diagnoses of AA and hypoplastic MDS or AML in cases where cytogenetics and molecular methods are not available at diagnosis (e.g., because of a culture failure). In some cases with pancytopenia, morphologically diagnosed AA with insufficient cytogenetics may progress to hematologic malignancies. Therefore, repeated follow-up, including bone marrow examination and cytogenetic analysis, is required to identify misdiagnosed cases. Moreover, SAA patients whose disease converts to non-severe AA, or vice versa, and who become independent of transfusion, need repeated examination of their bone marrow for the monitoring of potential evolution to other hematologic malignancies.
CAs are rarely seen in AA. The analysis of 127 AA patients suggests that, despite the small number of patients with CA in AA, CA did not affect the survival or evolution into other hematologic malignancies. As previously reported [8], adult non-severe AA patients may have CA at initial diagnosis or at follow-up. More patients and prospective studies are needed to establish the clinical significance of CA in AA.
Go to :
 XML Download
XML Download