

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Bone marrow transplantation (BMT) has emerged over the past 50 years as a life-saving therapy for many human diseases. Although the first reports of successful transplantation described children with inherited immune deficiency disorders, one with severe combined immunodeficiency (SCID) [1], and another with Wiskott-Aldrich syndrome (WAS) [2], most recipients of allogeneic hematopoietic stem cell transplant (HSCT) are adults and suffer from acquired disorders; leukemia, lymphoma, or aplastic anemia. Regardless of the underlying disease, from the beginning of HSCT, it became apparent that adoptive transfer of healthy marrow and the progeny of donor bone marrow derived hematopoietic stem cells can lead to full reconstitution of the immune system. Among the most significant advances in HSCT over the past decades have been explorations into the use of alternate hematopoietic grafts, to include allogeneic unrelated umbilical cord blood [3-5]. Cord blood (CB) is a by-product from childbirth that historically had been discarded. Experimental studies demonstrated that CB is enriched in primitive hematopoietic progenitors compared with adult bone marrow (BM) and mobilized peripheral blood stem cell grafts (PBSC), reviewed in [6]. Notably, long-term engrafting cells are almost eightfold enriched among CB CD34+ cells compared to those in CD34+ cells from PBSC. Moreover, CB-derived CD34+ progenitors display a ~15-fold higher multilineage proliferative capacity [7]. These biological features could explain the unexpected clinical finding that despite a slower pace of myeloid engraftment after CB transplantation there is a higher prevalence of early progenitors in the marrow space in those children who were given CB as opposed to BM transplant [8].
CB as a hematopoietic graft was first used in 1989 for a child with severe Fanconi anemia [9]. Since 1993, when the first ever unrelated cord blood transplant (UCBT) was performed at Duke University [10], >20,000 such transplants are estimated to have been performed worldwide [11]. The increasing use of CB in the unrelated donor setting is partially explained by the following significant findings. UCBT apparently offers a similar [12] or reduced risk [13] of severe graft-versus-host-disease (GVHD) permitting a less stringent criteria for HLA matching, reviewed in [14].
Although UCBT is a life-saving form of HSCT, it is limited by the high incidence of opportunistic infections (OI), most of which are viral. OI is the major cause of transplant-related mortality during the first 6 months after UCBT, and is caused by delays in immune reconstitution. Immune reconstitution is a highly complex process influenced by both graft (e.g. cell dose, histocompatibility, donor serology) and recipient-related factors (age, previous therapy, conditioning regimen, past infectious exposures, etc). Adaptive immunity is the cumulative effect of a network of phenotypically distinct and highly specialized leukocyte subpopulations. Antigen presenting dendritic cell (DC), effector, memory, and regulatory lymphocyte subsets are the principal players of this network. Instructional signals from the innate immune system shape the quality and net effect of antigen-specific immunity.
While attaining normal ranges of specific leukocyte subsets have been used extensively in the literature, protection from clinical infections is the best surrogate marker of successful immune reconstitution.
THE HIGH INCIDENCE OF INFECTIONS AFTER UCBT OCCURS PREDOMINANTLY WITHIN THE FIRST 3-4 MONTHS AFTER TRANSPLANT
Infection-related mortality (IRM) is the primary or secondary cause of death (with or without another major cause such as GVHD) in ≥50% of deaths after UCBT with the majority of them occurring in the first 100 days [14-17]. A classic report from the International Bone Marrow Transplant Registry (IBMTR) highlights the unique features of infection incidence after UCBT. Outcome after transplantation was analyzed between recipients of either CB (N=150) or from marrow that was from HLA-matched (N=367) or mismatched for 1 HLA antigen (N=83) [18]. IRM within 100 days after transplantation was significantly higher among recipients of mismatched CB than among recipients of either HLA-matched marrow or mismatched marrow (45%, 21%, and 24%, respectively; P=0.01). However, beyond day 100, the proportions of infection-related deaths were similar in the 3 groups. A multicenter study from Spain also found increased occurrence of severe infections during the first 100 days after UCBT (73%) than in the BMT/PBSC groups (50%; P=0.02) [19]. Similarly, the Minnesota group also documented higher cumulative incidence of serious infections among children receiving UCBT (58%) immediately post-transplant in contrast to recipients of unmanipulated BM (35%, P=0.04) [20]. However, subsequently there was a trend towards less serious infection in the UCBT group similar to the findings of IBMTR report [18], suggesting that the immune deficit that seems to be so heightened in the immediate post-UCBT period is followed by significant improvements of immunity. Although causal relationship cannot be established directly, this period coincides with the time of thymic recovery. When the Minnesota group analyzed 2 BMT cohorts separately, patients in the T cell-depleted group demonstrated significantly more viral (P<0.01), but not bacterial, infections than either UCBT or unmodified BM recipients beyond 6 months. These findings together with the report by the COBLT investigators [21] suggest an important protective role for post-thymic T cells infused in the graft regardless whether they include antigen experienced (marrow) or almost exclusively antigen naïve (CB) T cells.
GENERAL PRINCIPLES OF IMMUNE RECOVERY AFTER HCT
Immunoablation is an obligatory consequence of most myeloablative preparative regimens. While as a whole the innate immune system appears to recover rapidly, within weeks after HCT, the recovery of functional B and T lymphocytes (adaptive immunity) is far more difficult to achieve [22, 23]. The first wave of T cells emerging in the lymphopenic host are peripherally expanding T lymphocytes that were infused with the graft representing the thymic-independent pathway [24]. However, the antigen-driven expansion of peripheral T cells leads to a limited and skewed T cell receptor (TCR) repertoire [24]. Several weeks/months later a second wave of T cells emerge developing from donor-derived common lymphocyte progenitors (CLP) as the result of de novo thymopoiesis. In the absence of significant GVHD, this thymic-dependent pathway [25, 26] is solely responsible for a fully diverse T cell repertoire. Interestingly, by 2 years after HCT higher TCR diversity may be attained in CB recipients than in recipients of BMT [27] indicating the existence of an efficient thymic-dependent pathway.
PLACENTAL FACTORS SHAPE THE FUNCTIONAL PROFILE OF FETAL T CELLS AND DENDRITIC CELLS RESULTING IN FUNCTIONAL DEFICITS
CB contains significantly higher absolute numbers of T, NK, and B lymphocytes than adult peripheral blood (PB) or even marrow [28-31]. However, functional differences are more critical compared to numerical ones. Despite an overlapping CD45RA+/CD28+/CD27+ phenotype, CB T cells are fundamentally different from 'naïve' adult T cells mostly because they demonstrate a relative Th2/Tc2 bias. Multiple tiers of Th1/Tc1 suppressing factors exist at the maternal-fetal interface; IL-10 secreted by cytotrophoblasts [32], reduced local Tryptophan levels [33], increased progesterone levels [34] and other placental factors (IL-4, PGE2), reviewed in [35]. This immunoregulatory network could be viewed as part of an evolutionary adaptation to permit survival and avoid rejection of the fetus. Fas-ligand expression at the maternal-fetal interface may eliminate T cells activated despite these factors above [36]. The exuberant production of IL-13 primarily by CD8+ T cells [37] is in sharp contrast with lower IFNγ production that persists even after stimulation via CD3/CD28 signaling and exogenous IL-2 [37]. This is a consequence of differential patterns of methylation of the IFNγ promoter [38]. Independently, impaired APC function of neonatal/CB dendritic cells (DC) restricts the potential for optimal Th1 cell responses in neonates due to their low IL-12 expression [39-41]. Despite these limits, there is evidence that intrauterine viral infections (e.g. cytomegalovirus, CMV) could generate partial Th1 immune responses [35], though persistent and selective deficiency of antiviral Th1 CD4+ T cell is documented into early childhood [42]. The reduced capacity of bulk CB mononuclear cells to secrete cytokines and lymphokines has been reported to affect GM-CSF, M-CSF, IL-4, IL-8, IL-12, IL-15, and IL-18, reviewed in [43, 44]. There is reduced expression of NFATc2 (nuclear factor of activated T cells c2), a critical transcription factor necessary for up-regulation of these and other cytokines known to amplify T-cell responses. The relative cytolytic deficiency of CB T cells is associated with absent expression of granzymes and perforin [45], essential for the control of viral and other pathogens. Nevertheless, as Kyung-Duk Park in my laboratory demonstrated [46], in vitro priming against CMV is feasible, if Th1/Tc1 skewing cytokines influence CB T cells before and during antigen encounter. CD25+ T cells in CB are naturally-occurring regulatory T cells [47] with potent suppressor function as opposed to peripherally activated CD25+ cells [48]. Fewer UCB T cells display HLA-DR and CCR-5 activation markers, while the CD8+/CD57+/CD28- and CD8+/CD45RA+/CD27- 'cytotoxic', along with the 'skin homing' CLA+ T cell subsets are absent altogether [30]. Compared with adult blood more CB T cells progress through cell cycle and enter apoptosis. However, unlike in adult PB the majority of proliferating Ki-67+ T cells in UCB retain a CD45RA+/RO-, CD69-, CD25-, HLA- DR- 'resting' phenotype [30, 49]. Unlike in adult blood, there is also significant expression of telomerase in CB T cells [49]. In contrast with T cells, CB NK cells are functionally "mature" with comparable or better lytic activity than their BM-derived counterparts [50]. Not surprisingly, 'naïve' B lymphocytes are in excess in CB with an abundance of CD5+ B1 cells and CD23- immature B cells [30, 51].
PREVIOUSLY REPORTED FEATURES OF IMMUNE RECOVERY AFTER UCBT
Although mitogenic proliferative responses may already reach normal range in children 6-9 months after UCBT, T cell reconstitution is gradual and typically does not reach age-appropriate numbers before 9 months. Meanwhile, T cell recovery in adults typically extends beyond the first year, presumably related to the inferior output of TREC+ naïve T cells in older recipients [52]. Notably, NK cell recovery is prompt both in numbers and function in both adults and children by the first 2 months similar to recipients of BM [53-55]. Significant B lymphocyte recovery starts ~2-4 months after transplant and according to a recent analysis of a combined dataset from Marseille and Lyon, they may recover relatively fast, by ~3 months after CBT compared to ~6 months post-unrelated donor BMT [55].
Although the incidence of life-threatening viral infections is high in the first 6 months after UCBT likely reflecting deficits in T cell numbers or function, when monitored beyond 9 months post-transplant the speed of T cell recovery seems to be at least comparable [56] to or even better than that seen after unrelated BMT, [31, 53, 57]. Investigators from the Cord Blood Transplantation Study (COBLT) analyzed antigen-specific proliferation after UCBT [21]. Children with malignancies were longitudinally tested over the first 3 years post transplant for herpes virus specific responses (HSV, VZV, CMV). Approximately 43% of the patients studied eventually developed a positive T-lymphocyte proliferative response to at least one herpes virus at some point over the 3 year observational period. In a few, proliferative responses developed as early as within the first 30-50 days, indicating that naïve T lymphocytes transferred in the graft can give rise to antigen-specific T-lymphocyte immunity before thymic recovery [21]. Surprisingly, patients with a proliferative response at any time in the first 3 years to any of the herpes viruses had a lower probability of leukemia relapse and a higher overall survival [21]. One may speculate that the superior proliferative T cell response represents a powerful surrogate marker for functional immune reconstitution leading to more effective graft-versus-leukemia (GVL) activity. However, the development and kinetics of protective antigen-specific function was not evaluable [58].
Investigators at the University of Minnesota retrospectively evaluated the impact of overall lymphocyte recovery in 360 consecutive patients with hematologic malignancy utilizing data from standard hemocytometers [59]. Patients underwent UCBT between 2001 and 2007. In multivariate analysis, an absolute lymphocyte count (ALC) of >200×106/L at day 30 (N=73) after myeloablative conditioning was associated with superior 2-year overall survival (OS) (73% vs. 61%; P=0.02), progression-free survival (PFS) (68% vs. 54%; P=0.05) and reduced transplant related mortality (TRM) (8% vs. 28%, P<0.01) compared to those whose ALC was ≤ 200×106/L (N=43). Similarly, an ALC>200×106/L at day 42 (N=105) after RIC was associated with superior 2-year OS (59% vs. 41%, P<0.01) compared to those below (N=55). There was no significant relationship between ALC and relapse [59].
PATIENT AND GRAFT SPECIFIC FACTORS PREDICT THE RISK OF DEATH FROM OPPORTUNISTIC INFECTIONS IN THE FIRST 6 MONTHS AFTER UCBT
Over the past several years the Szabolcs lab has studied the reconstitution of immunity in the immediate post-UCBT period (prior to thymic recovery) in >150 pediatric recipients of single unit UCB at Duke University to identify surrogate immune markers for those at risk for opportunistic infections (OI).
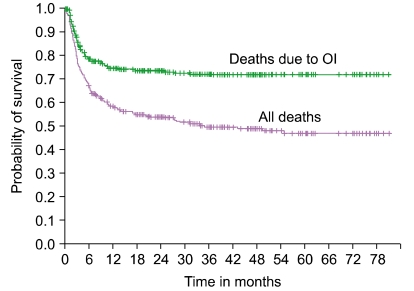
Several graft and patient-specific variables were identified as significant factors, when the laboratory measurements of DC and T cell reconstitution were analyzed. To determine the impact of patient and graft-specific factors on 6-month post-UCBT OI-related mortality, we reviewed all consecutive pediatric UCB recipients transplanted at Duke University Medical Center between June 1999 and Oct 2005 to overlap with the immune monitoring studies [60]. Three hundred thirty pediatric recipients of single UCB grafts were identified. Those receiving a second transplant for primary graft failure were excluded. Two hundred twenty of the 330 patients (67%) were alive at 6 months (Fig. 1). Of those who died by 6 months, 58% were identified with OI (viral, fungal, protozoal infections) implicated as a cause of death. Those who died prior to 6 months and for whom OI was not implicated as a cause of death were omitted from the study dataset, resulting in 284 patients.
Multivariate modeling revealed that a significantly greater probability of 6-month OI-related death was associated with CMV-positive serology, greater HLA mismatch, and older age. Higher total graft cell dose, including CD34+ progenitor cell dose and CD3+ cell dose were each associated with lower probability of death due to OI at 6 months [61]. Here we demonstrated for the first time the protective immunity afforded by expansion and functional contribution of post-thymic T cells infused with the graft prior to the recovery of the "central" de novo thymic pathway. We are in the midst of analysis of longitudinal monitoring of dendritic cell and lymphocyte recovery during the first 2 years after UCBT with emerging protecting influence of Tregs in the first 6 months coupled with the protective effects of thymus regeneration at the 6 months time point and CD123+ plasmacytoid dendritic cell recovery, data not shown.
CIRCULATING DENDRITIC AND T CELL SUBSETS AT DAY +50 AFTER UCBT SERVE AS SURROGATE MARKERS OF PROTECTIVE IMMUNITY
To identify patients who were at increased risk for developing OI in the first 100 days, a prospective cross-sectional study has been conducted at day +50 post-UCBT [62], with the latest analysis extended to 111 patients. Utilizing Trucount™ methodology, 4-color surface and intracellular (ic) FACS was employed to accurately enumerate and characterize lymphocyte and DC subsets [30, 63, 64]. All patients received myeloablative conditioning regimes (TBI/CY, Bu/CY, Bu/MEL, TBI/MEL) and equine ATG at 30 mg/kg/day between day -3 to day -1. All received identical GVHD prophylaxis consisting of Cyclosporine A plus steroids, slowly tapered after day +21 in the absence of ≥grade II aGVHD. Various degree of cellular reconstitution is noted for most immune cells except for the absence of B lymphocytes. However, immune reconstitution varied widely.
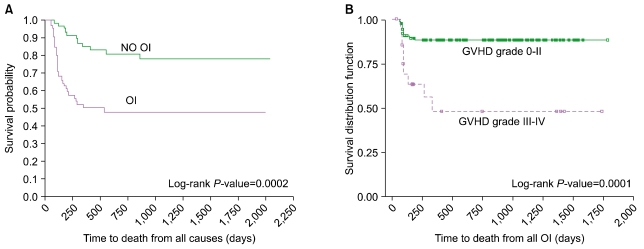
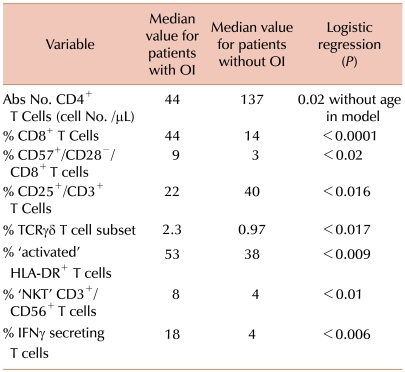
Table 1 lists those immune parameters that remain significant predictors for the presence of de novo developed OI. Fig. 2 shows that individuals that develop OI by day +100 have a significantly reduced probability of overall survival (Fig. 2A) and that death due to OI is related to Grade III/IV GVHD (Fig. 2B). Based on these data [62], and also on data not shown, we hypothesize that the increased prevalence of CD8+ T cells expressing/secreting HLA-DR, IFN, Granzymes A, B, Perforin represent an effort by the emerging immune system to control the infectious agent. These changes accompany down regulation of CD28 and CD27 expressions along with CD57 upregulation thus represent an evolution towards effector phenotype and function. Along with the skewing of the T cell profile, significantly fewer CD123+ plasmacytoid/lymphoid DC circulate in those with infection (P=0.007) demonstrating that antigen-presenting cell deficiency occurs along with lymphocyte alterations.
TH1 AND TC1 T CELL SUBSETS MEASURED AT DAY +20 AFTER UCBT CAN PREDICT THOSE AT RISK FOR OI
With another study we aimed to gain insight into the fate and maturational biology of adoptively transferred naive T cells in the lymphopenic hosts even prior to the onset of OI to develop predictive models for OI incidence in the first 100 days. Blood was obtained at a median 18 days post-UCBT if the WBC exceeded 400/mm3. Circulating T-cell subsets and DC counts were monitored. Since our last report [65], we have analyzed 76 patients at a median age of 62 months with at least 12 months follow-up. Forty four patients (58%) presented de novo with OI (>90% viral) at a median of 35 days. Both the OI+ and OI- patient cohorts had low but equivalent absolute WBC, CD3+, CD4+ T cells, and NK lymphocytes. DC subsets were largely undetectable. Strikingly, ~40% of circulating T cells were proliferating (Ki-67+), regardless of OI status, reflecting vigorous peripheral expansion reducing the CD45RA+/CD62L+ RTE pool to <20% from >90% infused in the graft only 2-3 weeks earlier [30]. While most cells (67±27%) expressed a 'memory like' CD45RA-/CD45RO+ phenotype, a significant population (14±28%) co-expressed CD45RA and RO. The robust T cell expansion was accompanied by upregulation of markers of activation with a median of 66% of T cells HLA-DR+. Interestingly, ~10% of the circulating T cells were entering apoptosis (ic activated Caspase-3+), regardless of OI status.
In those who developed OI, significantly higher proportion of the circulating T cells were CD8+ (40% vs. 28%, P=0.04), expressed CCR-5 (82% vs. 55%, P=0.009), were secreting IFN (35% vs. 12%, P=0.01), and acquired a CD57+/CD28- 'effector CTL' phenotype. In patients developing OI significantly more Perforin+/CD8+ T cells circulated (48% vs. 26%, P=0.02).
In conclusion, in the immediate post-transplant lymphopenic period extensive T cell proliferation via peripheral expansion leads to major immunophenotypic alterations accompanied by a gradual loss of the original naïve phenotype. In parallel, new T cell subsets emerge displaying a phenotype associated with antigenic stimulation [66]. We hypothesize that in patients who will develop OI, even clinically undetectable levels of virus could induce phenotypic acquisition of Th1/Tc1 cytotoxic effector profile.
CORD BLOOD T CELL EXPANSION IN VITRO WITH TH1 TC1 MATURATION; PRECLINICAL RESEARCH TOWARDS ADOPTIVE T CELL IMMUNOTHERAPY
Recently, we and others have demonstrated the feasibility of ex vivo CB T cell expansion [67, 68], drawing from the pioneering work by June et al. [69] utilizing paramagnetic Dynal beads coated with anti-CD3 and anti-CD28 stimulatory antibodies. These artificial antigen presenting cells (APC) simultaneously provide agonistic TCR and co-stimulatory signals triggering sufficient T cell proliferation in vitro to generate clinically relevant DLI products from living donors [70-72]. Although in our previous work [67], robust T cell expansion and even partial Th1/Tc1 maturation was evident starting with frozen/thawed CB specimens, significant apoptosis (~16%) resulted in an inverted CD4/CD8 ratio and diminished yield despite relatively low concentrations of IL-2 in the medium. The high degree of apoptosis was likely the result of activation induced cell death (AICD) following strong TCR signaling on CB T cells as previously described [73]. Infusion of overstimulated, apoptosis prone DLI product would likely lead to a narrow T cell repertoire and shortened T cell survival in vivo. Moreover, it could falsely suggest futility of ex vivo expanded DLI strategies.
In the most current study [74], we tested and confirmed our hypotheses, that interleukin-7 (IL-7) acting in concert with a new, clinical grade CD3/CD28 costimulatory bead and IL-2, would not only enhance ex vivo CB T cell proliferation while retaining a broad TCR repertoire as predicted [75], but it would also reduce activation induced cell death (AICD).
FAVORABLE IMPACT OF IL-7 ON CB T CELL SURVIVAL, PROLIFERATION, AND TCR V REPERTOIRE DURING CD3/28 MEDIATED EXPANSION
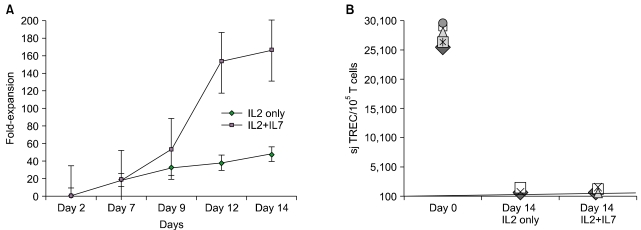
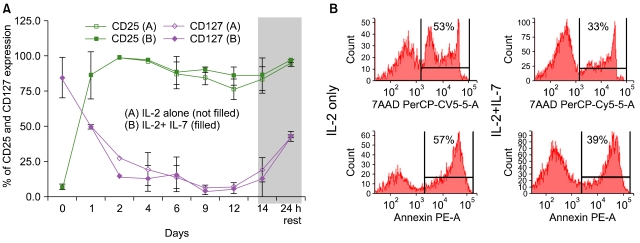
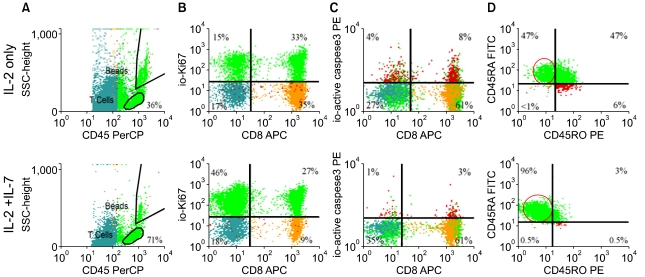
Purified T cells obtained from frozen/thawed cord blood specimens were split and cultured in parallel with and without IL-7. Matched pair analysis demonstrated significantly more viable T cells when IL-7 was added to IL-2 in the medium leading to an average of 165 fold T cell expansion (Fig. 3A). Following 14 days of expansion, striking dilution of TCR excision circles was noted as the sjTREC content in CD3+ T cells was depleted by ~2log in both culture conditions as compared to the starting population of pre-expansion CB T cells (Fig. 3B), irrespective of IL-7 exposure. Significantly more viable CD45 bright T lymphocytes were identified in cultures supplanted with IL-7 (71±10%) compared to cultures with IL-2 alone (46±15%) As determined by ic activated Caspase-3 expression and 7-AAD staining, there were significantly fewer T cells undergoing active apoptosis in the presence of IL-7 (median 4% versus 8%) (Fig. 4B). The anti-apoptotic effect of IL-7 was evident in both CD4+ and CD8+ subsets. To test T cell survival promoting effects of IL-7 beyond the in vitro expansion period, expanded cells were frozen on Day 14 and subsequently thawed and rested for 24 h in culture medium devoid of cytokines. Although the rest period in vitro can not mimic the in vivo post-infusion conditions exactly, these experiments demonstrate that T cells post expansion retain the potential to up-regulate IL-7 receptor/CD127 (Fig. 4A), and that the majority of IL-7 + IL-2 expanded T cells are still alive after freeze, thaw, and 24 hour culture in medium (Fig. 5B). Independent of the described anti-apoptotic effects, IL-7 promoted significantly greater T cell proliferation. About 2/3rd of all T cells were still actively cycling at the termination of the expansion period, as detected by intracellular Ki-67 expression (Fig. 5). Since naïve T cells with recent thymic emigrant phenotype (CD28+/CD27+CD45RA+/CD62L+) represent the vast majority of unmanipulated CB T cells, these findings corroborate earlier studies demonstrating the proliferative and anti-apoptotic effects of IL-7 to be operational predominantly in the naïve/CD45RA+ T cell compartment [75, 76]. In addition to superior T cell proliferation and reduced apoptosis in IL-7 supplanted conditions, we also found higher TCRVβ diversity per family (P=0.04, N=3) displaying a broad polyclonal spectrum [74] (data not shown).
LIMITED TH1/TC1 'MATURATION' DURING EXPANSION AND LOW EXPRESSION LEVELS OF 4-1BB/CD137, CD40L, AND PERFORIN CORRELATE WITH ABSENT ALLOREACTIVITY
Once we have demonstrated the salutary effects of IL-7 on T cell viability, expansion, and overall T cell receptor diversity, we sought to determine its impact on surface and intracellular phenotype and overall T cell function as measured by cytokine secretion profile and cytotoxicity. Despite undergoing several cycles of cell division triggered by IL-2 + IL-7 in concert with TCR and CD28 co-stimulation, significantly more CB T cells retained the naïve starting phenotype, CD45RA+/CD62L+ in the IL-7-containing condition (90±5%) compared to cells cultured in IL-2 alone (73±14%, P=0.03) (Fig. 5). Surface expression of L-selectin (CD62L) is essential for effective T cell homing to secondary lymphoid organs, a desired destination for antigen inexperienced, unprimed adoptive T cell infusions. CCR-7, a chemokine receptor implicated in both the entry and also in the retaining of T cells in lymph nodes, was also expressed on the majority of expanded T cells, data not shown. Interestingly, while the surface of post-expansion T cells appeared identical to unmanipulated fresh cord blood T cells in terms of CD28+/CD27+/CD45RA+/CD62L co-expression, expanded T cells displayed several upregulated surface molecules commonly seen after activation, including CD25, HLA-DR, OX40. However, <10% of cells expressed CD40L. Despite the preservation of resting, naïve, 'RTE-mimicking' surface phenotype, as indicated by CD28+/CD27+/CD45RA+/CD62L co-expression, CD3/CD28 co-stimulation led to rapid down-regulation of membrane CD127 (IL7Rα) in parallel with surface CD25 (IL2Rα) up-regulation on the very same T cells (Fig. 4A). This "receptor switching process' is not dependent on the presence of IL-7 in our cultures as CD25 and CD127 expression levels were superimposable in the presence and absence of IL-7 (Fig. 4A). Moreover, since a near complete reversal between CD25 and CD127 expression has occurred by ~24-48 h of culture this phenomenon appears independent of cell division. Numerical T cell expansion does not begin in earnest in the cultures until day-3-4 (Fig. 3). Interestingly, when expanded T cells were frozen on Day 14 and subsequently thawed and rested for 24 h in culture medium devoid of cytokines, CD127 was re-expressed on nearly half of the viable T cells (Fig. 4A). IL-7 receptor re-expression could permit delivery of pro-survival signals to expanded T cells administered by clinical DLI infusions in the lymphopenic post-transplant state where endogenous IL-7 level has been demonstrated to be elevated up to 10-30 pg/mL weeks after transplant [77]. Although it is possible that high levels of IL-7 in vivo could induce down-regulation of CD127 in the responding T cells, nevertheless our results suggest that the expanded T cells retain the capacity to re-express CD127 even when rested post-thaw with IL-7 at 15-30 pg/mL (data not shown).
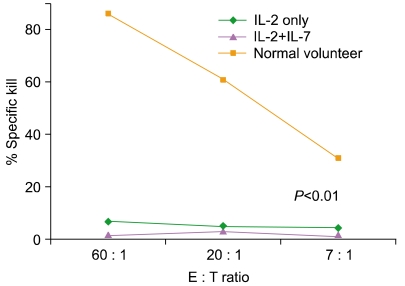
CD3/CD28 co-stimulation with ClinExVivo™ Dynabeads® in this series of experiments enhanced in a larger fraction of post-expansion T cells the capacity for intracellular expression of IFNγ, TNFα, and Granzyme B than we previously reported using different artificial-APC beads [67]. Nevertheless, despite the potential for an increase in alloreactivity [78] after the more robust expansion in the presence of IL-7, the expanded progeny lacked cytotoxicity against a highly immunogenic (CD40+, CD80+, CD86+) EBV+ allogeneic lymphoblastoid cell line (IM9) (N=7), or recipient fibroblasts (N=2), despite a week long pre-sensitization prior to performing the CTL assay (Fig. 6). Interestingly, absent cytotoxicity coincided with low expression of 4-1BB/CD137, CD40L, and perforin. Taken together, these features support a favorable safety profile of 'day 14' ClinExVivo™ expanded T cells with reduced likelihood for inducing GVHD in vivo upon adoptive transfer.
EX VIVO EXPANDED, CD3/CD28-COSTIMULATED CORD BLOOD T CELLS CAN BE PRIMED IN VITRO AGAINST LYMPHOID AND MYELOID LEUKEMIA
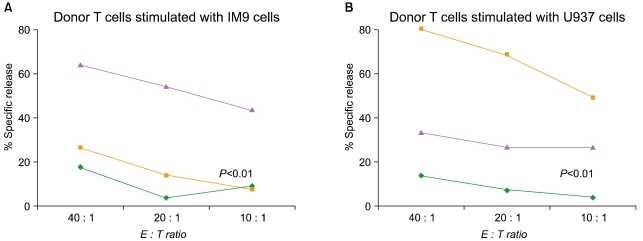
Donor leukocyte infusion with 'day 14' ClinExVivo™ +IL-2 +IL-7 expanded T cells generated from the originally infused CB graft could alleviate post-transplant lymphopenia and qualitative T cell defects until thymic regeneration could contribute new T cells. However, such DLI would be antigen non-specific and will require microbial and/or tumor antigens to in vivo prime infused T cells in the transplant recipients. In a series of experiments, we evaluated the potential of 'day 14' CD3/CD28-costimulated/expanded T cells to undergo in vitro priming against specific leukemic targets. In vitro generated tumor-specific CTL responses could be adoptively infused to treat leukemia patients with minimal residual disease and/or relapse. CD3/CD28-expanded 'day 14' T cells were stimulated in vitro for 3 weeks in parallel cultures with killed, Mitomycin C treated lymphoid leukemia cells (IM9) and IFNγ-treated myeloid leukemia cells (U937). U937 cells by themselves can not induce allogeneic T cell response unless a stimulating anti-CD3 antibody is added to cultures [79]. In addition, they do not provide co-stimulation via the CD80/CD86-CD28 pathway, but likely via the ubiquitously expressed CD147, CD98 molecules [79]. CTL priming was performed in the decpreasing presence of IL-12, IL7, and IL-15 drawing from our published experimental strategy to in vitro prime anti-viral responses from CB [46]. Robust T cell expansion (195X, ±115, N=4) ensued over the course of ~3 weeks when killed leukemia cells rather than ClinExVivo™ beads served as APC. After the course of 2 to 3 repeated stimulations, strong leukemia-specific cytotoxicity was detected in CTL assays, killing the stimulating leukemia cells but not the other leukemia or most importantly CB transplant recipient PHA blasts (N=4, P<0.01). (Fig. 7). Failure to recognize and kill CB transplant recipient PHA blasts indicates future clinical safety from the potential toxicity of GVHD.
CONCLUSION
UCBT is a life-saving form of HCT, however, it is limited by the high incidence of OI, most of which are viral. OI is the major cause of transplant related mortality during the first 6 months after UCBT, and is caused by delays in immune reconstitution. For several months, until recovery of the thymus is restored to support de novo T cell generation, protective antiviral immunity depends on the activity of post-thymic T cells that are infused within the CB grafts. However, almost all CB T cells are antigen inexperienced (naïve) lymphocytes that have been functionally altered by placental factors to protect pregnancy. CB T cells need to undergo in vivo priming, Th1/Tc1 maturation, and peripheral expansion before they can afford immunologic protection. Despite advances in our understanding of cellular immune recovery after UCBT, fundamental gaps in knowledge remain regarding the biology and kinetics of developing antigen-specific protective immunity and understanding the impact of recipient age. However, there is also realistic hope that novel immunotherapy strategies could enhance immune competence to reduce infectious morbidity, relapse, and improve survival after CBT.
 XML Download
XML Download