

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Therapy-related acute leukemia (t-AL) is the most severe complication that may occur after treatment for primary cancer. It is diagnosed on the basis of histories of treatment and molecular abnormalities, and develops after chemotherapy, radiation, or immunosuppressive agents performed for the management of the primary cancer [1]. The World Health Organization (WHO) recognizes therapy-related myeloid neoplasm (t-MN) as a distinct subtype of acute myeloid leukemia (AML), although acute lymphoblastic leukemia (ALL) may also arise [2]. Topoisomerase inhibitors and alkylating agents are 2 well-known antineoplastic agents that cause t-AL [3].
The incidence rate of t-AL among patients treated for breast cancer has been reported to be 0.2-0.5% [4]. Breast cancer is the second most common cancer in Korean women, and such serious side effects are a matter of concern [5]. Topoisomerase inhibitors are part of cocktail treatment regimens for breast cancer patients. Topoisomerase inhibitors act as antineoplastic agents by binding to the DNA/enzyme complex inhibiting the action of topoisomerase, thus leaving behind a single strand of DNA. Multiple DNA-strand breaks lead to cell death but these breaks, which are repaired by chromosomal translocation, initiate leukemogenesis [1].
t-AL constitutes 10-20% of all cases of AL [1]. However, only sporadic cases of t-AL after systemic chemotherapy have been reported in Korea [6]. Although many epidemiologic studies have been conducted in other countries, no such literature exists on t-AL in the Korean population. Herein, we report the clinical characteristics, chromosomal findings, and bone marrow test results of 12 cases of t-AL that developed after topoisomerase inhibitor therapy in breast cancer patients in a single institute.
Go to : 
MATERIALS AND METHODS
1. Case selection
We reviewed the electronic medical records of patients who were primarily diagnosed with breast cancer and later developed AL and underwent treatment at the Asan Medical Center, Seoul, Korea, from 1989 to October 2009. A total of 9,228 patients were diagnosed and treated for breast cancer. Among them, 14 patients (0.2%) later developed t-AL. Two patients were excluded from this study: the therapeutic regimen of 1 patient did not include topoisomerase inhibitors and the other patient had been treated at a different hospital, and her medication history could not be evaluated. We retrospectively evaluated the medication record, history of radiation treatment, bone marrow test results, immunophenotypic results, and cytogenetic results of the included patients.
2. Morphologic examination and immunophenotypic analysis
Peripheral blood and bone marrow aspirates were prepared with Wright-Giemsa staining for morphologic evaluation. Conventional panels were chosen for the analysis of AML and ALL markers and the FACSCanto flow cytometer (BectonDickinson, San Jose, CA, USA) was used.
3. Cytogenetic analysis
Cytogenetic analysis was performed on bone marrow specimens using standard techniques. Giemsa-trypsin-Giemsa (GTG) banding was used to identify the chromosome. At least 20 metaphase cells were examined after 24-hr culture. The chromosomes were evaluated under 300-400 bands resolution according to the International System for Human Cytogenetic Nomenclature (2009) [7].
4. Statistical analysis for event-free survival
Event-free survival of 12 patients was analyzed. We divided the cases into the 11q23 rearranged group and non-11q23-involved group. The Wilcoxon test was used to plot the survival graph of the 2 groups.
Go to :
RESULTS
1. Clinical features of the cases
Doxorubicin (Admycin; BoRyung Pharm, Seoul, Korea), which inhibits the progression of topoisomerase, was used in 12 patients. Alkylating agents were used together with doxorubicin in 10 patients. The participants were all female patients with a median age of 55 years. The period between completion of chemotherapy for breast cancer and diagnosis of t-AL was 18-95 months with a median period of 25 months. The patients' event-free survival after diagnosis of AL was 1-39 months. Five patients died, 3 patients are still alive, and 4 patients were lost to follow-up (Table 1).
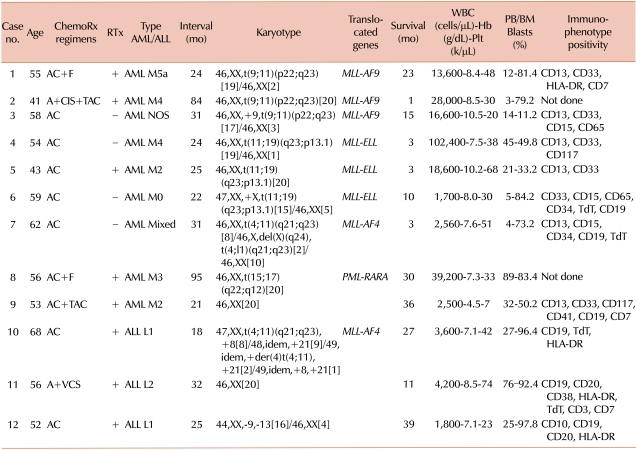
Table 1
Demographics and laboratory findings of 12 patients.
Abbreviations: A, Admycin (doxorubicin hydrochloride); C, Alkyloxan (cyclophosphamide); F, 5FU (fluorouracil); CIS, Cisplatin; TAC, Taxol (paclitaxel); VCS, vincristine sulfate; RTx, radiation treatment; AML, acute myeloid leukemia; ALL, acute lymphoblastic leukemia; TdT, terminal deoxynucleotidyl transferase; HLA, human leukocyte antigen.
![]()
2. Hematologic features of the cases
Nine patients were diagnosed with therapy-related AML (t-AML) (75%, 9/12) and 3 patients (25%, 3/12) with therapy-related ALL (t-ALL). At the time of diagnosis, the average white blood cell count was 13,066/µL; median platelet count, 33,000/µL; median hemoglobin level, 7.6 g/dL; median percentage of blasts in the blood, 21%; and median percentage of blasts in the bone marrow aspirates, 79.2%. Leukemic blasts showed a "packed marrow" pattern with cellularity ranging from 75 to 100%.
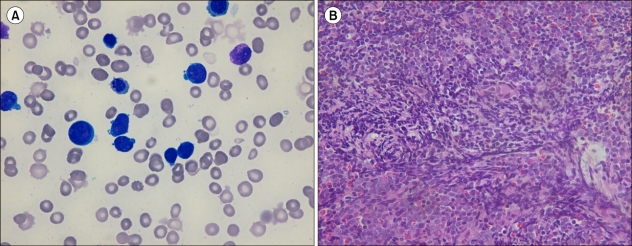
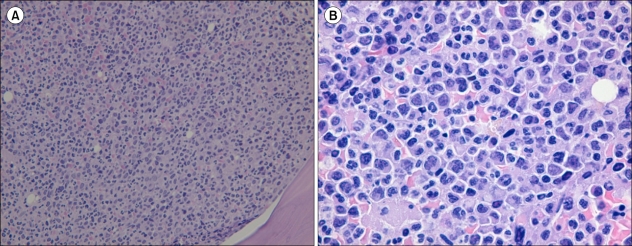
One patient with mixed-lineage AML (case No. 7) showed 2 distinct populations of blasts (Fig. 1A) in the marrow aspirates. Also, her bone marrow biopsy revealed extensive myelofibrosis involving many myeloblasts and lymphoblasts (Fig. 1B). In another patient (case No. 3) diagnosed with t-MN, the percentage of blasts in the bone marrow aspirate was only 11.2%, and her bone marrow biopsy showed "packed marrow" infiltration of myeloid cells with variable maturation stages and scattered myeloblasts (Fig. 2).
3. Cytogenetic features of the cases
Eight patients (67%, 8/12) had translocation of 11q23 (MLL), of which 3 patients showed translocation of 19p13.1 (ELL) (37.5%, 3/8), 3 patients of 9p22 (AF9) (37.5%, 3/8), and 2 patients of 4q21 (AF4) (25%, 2/8). The karyotypes of the remaining 4 patients were 2 normal karyotypes, 1 t(15;17), and 1 with -9/-13. Among the 8 cases of 11q23 rearrangement, 7 cases were diagnosed as t-AML and only 1 case was diagnosed as t-ALL.
4. Survival analysis and period between completion of treatment for breast cancer and diagnosis of t-AL; 11q23 vs. non-11q23
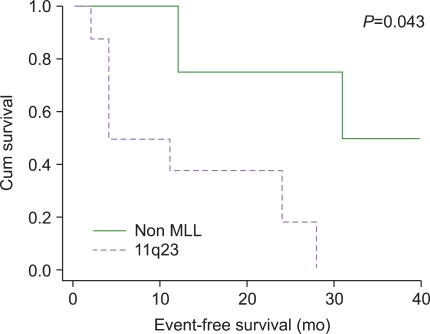
We analyzed the event-free survival of 12 patients. We divided the cases into 2 groups, the 11q23-rearranged group and the non-11q23-involved group. The Wilcoxon test was used to plot the survival graph of the 2 groups; the graph showed that the 11q23 group had significantly poorer survival (P=0.043) (Fig. 3). The mean period between completion of treatment for breast cancer and t-AL was 32.4 months in the 11q23 group and 43.3 months in the non-11q23 group (P=0.234).
Go to :
DISCUSSION
In this study, we selected cases with a history of treatment with a topoisomerase inhibitor prior to the onset of acute leukemia. Among the 12 patients treated with a topoisomerase inhibitor, 8 patients (67%) displayed a balanced translocation of the MLL gene located in the 11q23 chromosomal region. A balanced translocation involving 11q23 is characteristic of topoisomerase inhibitor-induced t-AL [8]. One patient who was excluded from this study because we did not have her medication record showed t(3;3), suggesting a lower likelihood of topoisomerase inhibitor usage. Except for 1 patient, the latency period between completion of chemotherapy and diagnosis of t-AL ranged from 18-32 months in 11q23-involved cases, which agreed with the established period of 1-3 years. In the patient with PML-RARA rearrangement (case No. 8), t-AL was diagnosed 95 months after completion of chemotherapy which was relatively long compared to the cases with MLL gene rearrangement. The latency period between completion of chemotherapy and diagnosis of t-AL for cases with PML-RARA rearrangement has not been reported but it is not thought to be longer than that of cases with MLL rearrangement. Although the latency periods are established in general, it can vary individually depending on many risk factors.
The survival analysis of our cases showed that t-AL with 11q23 rearrangement presented poor prognosis. t-AL cases with 11q23 rearrangements are known for poor prognosis and 1 study showed that only 80% of patients achieved remission and 75% of them relapsed within 1 year [9]. Chen et al. reviewed t-ALL cases reported in the literature and concluded that the prognosis of t-ALL was much poorer compared to t-AML [10]. They also noted that survival did not differ between the 11q23 group and non-11q23 group in t-ALL patients. The overall prognosis in t-ALL was poor, regardless of the presence or absence of 11q23 abnormalities. Although our cases are limited in number, 1 patient with 11q23-involved t-ALL (case No. 10) had a relatively long event-free survival (27 months) compared to the other 2 t-ALL patients.
The MLL protein transcribed by the MLL gene is a critical transcriptional regulator and is essential for the maintenance of adult hematopoietic stem cells and progenitors. A key feature of MLL fusion proteins is their ability to efficiently transform hematopoietic cells into leukemic stem cells [11]. Over 40 different partner genes for 11q23 have been reported, with 9p22, 19p13.1, and 4q21 being the most well-known [12]. The results of our cases agree with the previously reported data. Up to 30% of t(11;19)(q23;p13.1), 10% of t(9;11), and 5% of t(4;11) were found in patients with secondary leukemia. The gene located in the 9p22 chromosomal region is MLLT3, myeloid/lymphoid or mixed lineage leukemia translocated to 3, also known as AF9 (ALLL1-fused gene from chromosome 9), is responsible for proteins that act as transcription activator and is a frequent partner gene in MLL rearrangements. In de novo cases, the prognosis of t(9;11)(p22;q23) leukemia may not be as poor as in other 11q23 leukemia, but a very poor prognosis may be expected in patients with t-AL [13]. The 19p13.1 chromosomal region bears the ELL (eleven-nineteen lysine-rich leukemia) gene that transcribes proteins that function as RNA polymerase II elongation factor. t(11;19)(q23;p13.1) is related to very poor prognosis. A hybrid, mutated gene of 5'MLL-3'ELL produces abnormal proteins similar to other MLL fusion proteins and plays a role in oncogenesis [14]. AF4-MLL translocation, t(4;11)(q21;q23), is also known to be related to poor prognosis [15].
Our study revealed that the incidence of t-AL among patients treated for breast cancer was 0.2%. A German study reported a rate of 0.2-0.5% of t-AL among breast cancer patients [16]. Such incidence rate suggests that there might be specific predisposing factors for t-AL. Host-associated factors have been hypothesized, including heritable predisposition due to enzymatic polymorphisms in genes involved in DNA repair or drug metabolism [17, 18]. Polymorphisms in the gene of phase I enzymes, mainly the CYP family and phase II enzymes, are associated with decreased enzymatic activity in t-AL. Reduced activity of these enzymes and reduced detoxification result in increased production of activated metabolites and increased toxicity of chemotherapy [19]. Another mechanism of secondary leukemogenesis is DNA repair defects. Its susceptibility may be based on polymorphisms of DNA repair and synthesis, leading to incomplete repair and persistence of mutations [18].
Other than hereditary predispositions, Martin et al. suggested that younger age and advanced stage of breast cancer predispose t-AML because these patients require higher doses of chemotherapeutic agents leading to t-AML [20]. In addition, they speculated on the possibility that early-onset breast cancer may have a genetic predisposition to t-AL and questioned the role mutations in the BRCA1 and 2 genes might play in the development of t-AL. The hypothesis could not be applied in our study because the participants were middle-aged women. Praga et al. reviewed 9,796 breast cancer patients and reported that the cumulative probability of developing AML/myelodysplastic syndrome (MDS) was 0.37% in patients treated with standard regimens, whereas the probability increased to 4.97% in those treated with higher doses of topoisomerase inhibitors. The dosage of chemotherapeutic agents had a significant effect on the development of t-AL [21].
Several studies showed that combined radiotherapy and chemotherapy increased the risk of t-AL [22], whereas some suggested that limited-field radiation in the therapeutic dose range has very little or no effect on the risk of t-AL [1]. In our study, 8 patients had received radiation therapy and we did not observe a statistically significant effect on survival or latency period between completion of chemotherapy and diagnosis of t-AL.
t-AML represents 10-20% of all AML cases [23], whereas t-ALL accounts for 2.3% of all ALL cases [24]. t-ALL accounts for a much lower proportion of cases of secondary leukemia than t-AML. The incidence of t-AML (75%) was higher than that of t-ALL (25%) in our study. The WHO distinguishes t-MNs as a distinct category, although ALL may also arise secondary to cancer therapy. Some groups reported that t(4;11) is associated with t-ALL, whereas t(9;11) is generally found in t-AML [25]. One case (case No. 10) with t-ALL with 11q23 abnormality showed t(4;11) which is in agreement with the report. A review of the literature revealed that 11q23 aberration was not observed in nearly one third of reported cases of t-ALL treated with topoisomerase inhibitors [10]. This finding indicates that mechanisms other than 11q23 aberration may also contribute to the t-ALL induced by topoisomerase inhibitors.
In our study, 10 patients were treated with both a topoisomerase inhibitor and an alkylating agent. However, most cases presented with either 11q23 aberration or a short latency period between completion of chemotherapy and diagnosis of t-AL, i.e., patients with t-AL induced by a topoisomerase inhibitor. t-AL induced by alkylating agents generally presents with a longer latency period (5-7 years), preceding myelodysplastic features and frequent deletion of chromosomes 5, 5q, and/or 7, 7q [3]. The reason for the larger effect of topoisomerase inhibitors compared to alkylating agents remains unclear. Other factors such as dosage of each neoplastic agent might have had an effect.
Our presentation of 12 cases showed various features of t-AL after treatment with a topoisomerase inhibitor. We also observed that various types of AML were diagnosed other than myelomonocytic leukemia which is known to be the most prevalent type in AML associated with MLL gene rearrangement. t-AL patients with MLL gene rearrangement had a significantly poorer prognosis. t-AL, which is a serious complication of cancer treatment, should receive more attention, especially since the incidence of breast cancer is increasing in the Korean population. In addition, further studies are required to identify and establish risk factors other than the hypothesized predisposing factors of t-AL.
Go to :
 XML Download
XML Download