

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Arsenic trioxide (ATO, As2O3) is a traditional Chinese medicine that has been used to treat several diseases such as anemia, dyspepsia, and some tumors [1]. ATO promotes elimination of acute promyelocytic leukemia (APL) by the induction of apoptosis [2]. The apoptotic effect of ATO is not restricted to APL but is also observed in other malignant cells including those of non-APL acute myeloid leukemia, myeloma, chronic myeloid leukemia, esophageal and ovarian carcinomas, and neuroblastoma [3-5].
Hypoxia or low oxygen tension is present at inflammatory sites, solid tumors, and bone marrow. To adapt to hypoxia, cells express many types of genes that are related to angiogenesis, mobility, and glucose metabolism through the hypoxia-inducible factor-1α (HIF-1α) [6-8]. HIF-1α is degraded by ubiquitination in the proteosome under conditions of normal oxygen tension (normoxia) even though it is constantly expressed. However, under hypoxia, HIF-1α is stabilized because of the lack of oxygen, and bound to constitutively expressed HIF-1β (ARNT). The HIF-1α/β heterodimer is bound to DNA at a specific recognition site known as the hypoxia response element (HRE), which initiates transcription of target genes [9, 10]. HIF-1α can be also stabilized by iron chelators and metal ions such as Co2+, Ni2+, and Mn2+ even under normoxia. In this study, we used cobalt chloride (CoCl2), a well-established chemical inducer of HIF-1α, to induce hypoxia-like responses [11].
Leukemic cells originate from bone marrow, which acts as a physical barrier to anticancer drugs. In addition, the microenvironment of bone marrow transmits survival signals that mediate adhesion through integrins and CD44 to fibronectin and hyaluronan [12, 13]. Although these factors increase leukemic cell resistance in the bone marrow to chemotherapeutic drugs, the survival effect of hypoxic bone marrow microenvironment on leukemic cells is still not understood.
Many previous studies have primarily focused on the apoptotic effect of hypoxia that affects cell survival and differentiation [14, 15]. The anti-apoptotic effect of hypoxia on several cell types has also been assessed [16-18]. Here, we investigated the relationship between hypoxia (using a mimetic agent) and drug resistance in leukemic cells to elucidate the contribution of hypoxic bone marrow microenvironment to failure of chemotherapeutic treatment.
MATERIALS AND METHODS
1. Cell culture and reagents
Four human leukemic cell lines (HL-60, APL; U937, acute myeloid leukemia; CCRF-CEM, acute lymphoblastic leukemia; K562, chronic myelogenous leukemia) were purchased from the Korean Cell Line Bank (KCLB; Seoul, Korea). All cells were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA) supplemented with 10% FBS (Hyclone Laboratories, Inc., Logan, UT) and penicillin-streptomycin (Invitrogen). To induce expression of HIF-1α, cells were cultured with a cobalt chloride (CoCl2; Sigma, St. Louis, MO).
2. Cell proliferation assay: MTT assay
The cell proliferation rate was determined by measuring the absorbance of the MTT dye (3-[4,5-dimethylthiazol-2yl]-2,5-diphenyltetrazolium bromide; Sigma) in living cells. Cells were inoculated in each well of a 12- or 24-well tissue culture plate. After 3 d, the MTT solution was added to each well and the plates were incubated for 3 or 4 hr at 37℃. Supernatants were then removed, and 100 µL of 0.04 N HCl-isopropanol was added to solubilize the formazan crystal formed by succinate dehydrogenase activity in viable cells. The plates were shaken at room temperature for 30 min, and then the absorbance at 540 nm was measured on a ThermoMAX microplate reader (Molecular Devices, Sunnyvale, CA).
3. Detection of apoptosis
Apoptotic cell death was detected by an annexin V-FITC/PI apoptosis detection kit (BD Pharmingen, San Diego, CA) or a caspase-3 detection kit (BD Pharmingen) according to the manufacturer's instructions. For the annexin V-FITC/PI kit, after adding 5 µL annexin V-FITC and 5 µL propidium iodide (PI), cells were incubated for 15 min at room temperature in the dark. Flow cytometry analysis was carried out within 1 hr. For caspase-3 detection, cells were washed twice with cold PBS and then resuspended in a Cytofix/Cytoperm solution and incubated for 20 min on ice. Following washing with a Perm/Wash buffer, cells were resuspended in a Perm/Wash buffer containing an anti-active caspase-3 antibody and incubated for 30 min at room temperature. After washing with a Perm/Wash buffer, the cells were prepared for flow cytometry analysis.
4. Western blot analysis
Total proteins were extracted by Pro-Prep (InTron Inc., Seoul, Korea) supplemented with phosphatase inhibitors (Sigma) and then normalized with a Micro BCA reagent (Pierce, Rockford, IL). Protein samples (30 µg each) were run on a 10% SDS-PAGE gel and transferred to PROTRAN-NC (Whatman, Clifton, NJ). Blots were blocked with 5% skim milk in TBST (0.1% Tween 20 in Tris-Buffered Saline) at room temperature for 1 hr and then incubated with Bax, HSP70, HSP90, Bcl-2, ERK, phospho-ERK, p38, phospho-p38, α-tubulin (Cell Signaling Technology Inc., Danvers, MA), or HIF-1α (BD Pharmingen) antibodies in blocking buffer (1:1,000) at 4℃ overnight. After washing 3 times with TBST, blots were incubated with horseradish-peroxidase (HRP)-conjugated secondary antibodies in blocking buffer (1:5,000) at room temperature for 1 hr. Blots were then washed 5 times with TBST and developed with the Super-signal reagent (Pierce).
5. HIF-1α inhibition assay
HL-60 cells were pre-treated with cadmium chloride (20 µM, CdCl2; Sigma) for 12 hr then cultured for 48 hr with CoCl2. Cell viability was assessed by the MTT assay, and HIF-1α expression was checked by western blot analysis as described above. The experiments were repeated 3 separate times under each set of conditions.
RESULTS
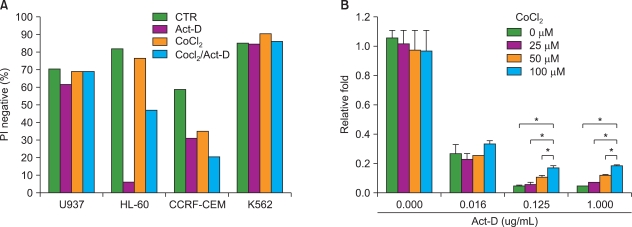
1. CoCl2 increases resistance of HL-60 cells to actinomycin-D
Leukemia cells originating from the bone marrow are under hypoxic conditions, which is in contrast to normoxic peripheral blood. We first investigated whether hypoxia-like conditions increased resistance of leukemic cells to actinomycin-D (Act-D)-induced oxidative stress-dependent apoptosis in U937, HL-60, CCRF-CEM, and K562 cells [19]. Among the 4 different leukemic cell lines, HL-60 cells were resistant to Act-D-induced cell death by treatment with CoCl2 (PI negative population: Act-D-treated, 5.8% vs. Act-D/CoCl2-treated, 47.09%) (Fig. 1). The protective effect of CoCl2 was significantly increased at higher doses of Act-D (0.125 and 1.0 µg/mL). These results indicate that CoCl2 increases resistance of HL-60 cells to Act-D-induced cell death.
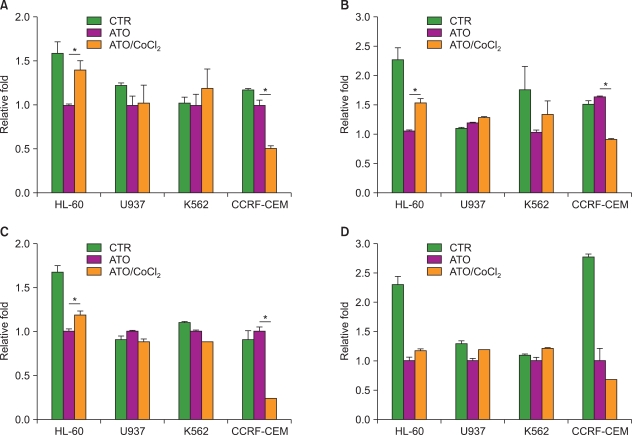
2. CoCl2 increases resistance of HL-60 cells to anti-leukemic drugs
Next, we tested the resistance of leukemic cells to several anti-leukemic drugs that are used for leukemia treatment because development of drug resistance is the most important factor associated with the failure of leukemic cell chemotherapeutic treatments. Each cell line was treated with an anti-leukemic drug: ATO (A), 17-(allylamino)-17-demethoxygeldanamycin (17-AAG), an HSP90 inhibitor (B), valproic acid (C), and cytarabine (D), in the presence or absence of CoCl2 (Fig. 2). Among the 4 cell lines, HL-60 cells were resistant under CoCl2-treatment conditions to each of the above anti-leukemic drugs (ATO, 1.41-fold increase, P=0.009; 17-AAG, 1.54-fold increase, P=0.002; valproic acid, 1.19-fold increase, P=0.007; cytarabine, 1.18-fold increase, P=0.053). K562 was resistant to only 17-AAG. In contrast, the treatment of CCRF-CEM cells with CoCl2 strongly decreased cell viability in the presence and absence of the anti-leukemic drugs (Fig. 1A and Fig. 2).
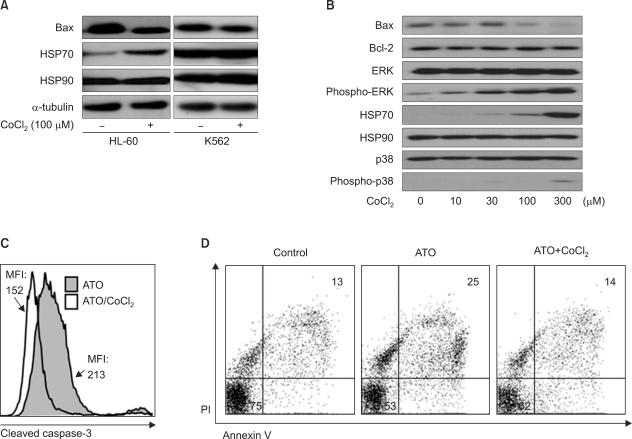
3. CoCl2 prevents apoptotic cell death in HL-60 cells via regulation of the apoptotic pathway
To verify whether hypoxia-like conditions inhibit HL-60 cell death by blocking the apoptotic pathway, we investigated the expression level of several apoptosis-related genes by western blot analysis based on previous results in 4 cell lines. Intriguingly, CoCl2 affects the expression of both the pro-apoptotic protein Bax (down-regulation) and the heat shock protein HSP70 (up-regulation), which are the main regulators of ATO-induced cell death [20, 21], but did not affect the expression of Bcl-2 or HSP90 (Fig. 3A, B). Moreover, we found that CoCl2 increased the phosphorylation levels of survival-related ERK and p38 (Fig. 3B). However, in K562 cells (Fig. 3A), U937, and CCRF-CEM cells (data not shown), there was no significant difference in the expression of Bax and HSP70 between CoCl2-treated and -untreated conditions. In addition, the level of cleaved caspase-3 was decreased with CoCl2 treatment in the presence of ATO, as compared to ATO in the absence of CoCl2 (Fig. 3C, mean fluorescence intensity (MFI); 152 vs. 213). Similarly, annexin V-FITC/PI analysis indicated that CoCl2 treatment decreased the apoptotic population of ATO-treated cells (Fig. 3D, annexin V/PI double positive: 25% vs. 15%). Collectively, these results demonstrate that CoCl2 treatment prevents apoptosis of ATO-treated HL-60 cells through the regulation of HSP70 and Bax.
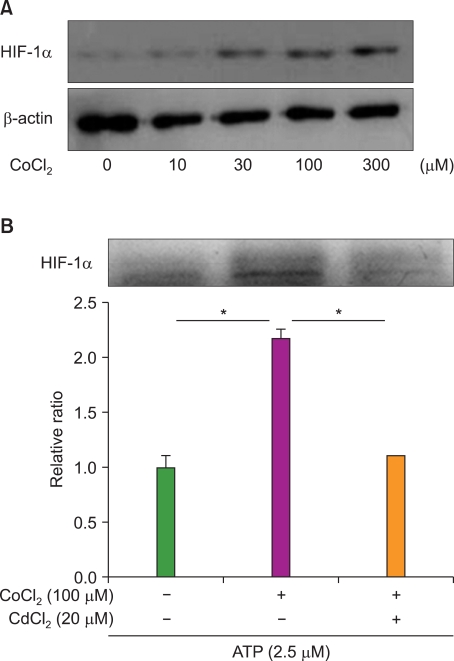
4. HIF-1α induction by CoCl2 increases the survival rate of HL-60 cells
As shown in Fig. 4, CoCl2 strongly increased the expression level of HIF-1α in a CoCl2-dose dependent manner (Fig. 4A) without significant cell death (data not shown). To determine if the effect of CoCl2 on decreased apoptosis was mediated by HIF-1α, we performed an inhibition assay using CdCl2, an inhibitor of HIF-1α activation [22]. CdCl2 effectively decreased the expression level of HIF-1α (Fig. 4B). While resistance of HL-60 cells was significantly increased more than 2-fold by CoCl2, treatment with CdCl2 reduced the survival rate of ATO-treated cells. These results show that the induction of HIF-1α by CoCl2 increases resistance of HL-60 cells to ATO.
DISCUSSION
Under hypoxia, the expression of migration and angiogenesis-related genes can be an important survival strategy in solid cancer cells. In contrast, some cells like immune cells in inflammatory sites and hematopoietic progenitor cells in bone marrow stay in hypoxic sites. In particular, the bone marrow provides both a physical barrier and survival signals to resident leukemic cells [12, 13]. Therefore, its protective effect increases resistance to anti-leukemic drugs. However, the effect of the hypoxic bone marrow micro-environment on leukemic cell survival is still unknown, although we previously reported that hypoxia decreases the migration rate of leukemia cells [23]. In this study, we examined the relationship between leukemic cell survival and HIF-1α, which is an essential effector of hypoxia.
First, we determined that CoCl2 treatment dramatically increased resistance of HL-60 cells to Act-D, an inducer of apoptosis, in a dose-dependent manner. Although the survival rate of HL-60 cells was not high due to the potent effect of Act-D, CoCl2 treatment strongly increased the PI negative population more than 7-fold. We then examined the effect of CoCl2 on cells treated with anti-leukemic drugs. CoCl2 increased resistance of HL-60 cells to ATO, 17-AAG, valproic acid, and cytarabine. However, we did not observe a protective effect of CoCl2 on other leukemia cell lines. For example, CCRF-CEM cells were relatively sensitive to CoCl2 treatment. These observations indicate that the anti-apoptotic effect of CoCl2 depends on the specific leukemic cell type.
ATO is currently used for acute promyeloid leukemia therapy due to its potential as an inducer of cell death through regulation of apoptosis-related molecules [2]. Results from the annexin V/PI analysis and caspase-3 activation assay indicated that CoCl2 treatment reversed the ATO-induced apoptotic rate of acute promyelocytic HL-60 cells. Although it is known that ATO induces cell death through the regulation of HSP70, HSP90, Bax, and Bcl-2 molecules [20, 21], our results indicate that CoCl2 regulates only HSP70 and Bax in ATO-treated HL-60 cells. Because the HSP70/Bax axis plays a critical role in the intrinsic apoptotic pathway [24, 25], its regulation by CoCl2 can be a potential mechanism to explain our observations. CoCl2 treatment also results in the phosphorylation and therefore activation of ERK and p38, which are related to increased survival of leukemic cells [26]. Data from the HIF-1α inhibition assay indicates that the survival effects of CoCl2 on ATO-treated HL-60 cells are strongly correlated with the expression of HIF-1α.
In some patients with leukemia, after initial chemotherapy, a small number of leukemic cells survive, develop resistance to treatment, and the disease returns. The most common relapse site of leukemia is the bone marrow where the leukemia originated. In addition to survival benefits provided by the bone marrow because of transmission of several survival factors to leukemic cells, such as cytokines and adhesion molecules [12, 13, 27], our results indicate that the hypoxic microenvironment of bone marrow may be a critical survival factor for leukemic cells. These discoveries provide new insights for understanding the mechanisms underlying hypoxia and for designing new therapeutic strategies for acute myeloid leukemia.
 XML Download
XML Download