

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cellular therapies are becoming increasingly important in treating cancer, hematologic malignancies, autoimmune disorders, and damaged tissue. These therapies include hematopoietic stem cell transplantation (HSC), adoptive immune therapy for cancer, gene therapy for the treatment of inherited immune deficiencies, and bone marrow stromal cell (BMSC) therapy for the treatment of ischemic heart disease. The contemporary cellular therapies now used involve the processes of cell selection, stimulation, culture, expansion, and gene insertion. Many even involve a combination of these processes. But while these therapies are becoming more effective and are being used more frequently, they are also becoming more complex.
HSC transplants have been used to treat acute leukemia and lymphoma for more than 40 years. Initially, this therapy involved the transplantation of marrow aspirated from syngeneic or HLA-compatible sibling donors. HSC transplantation has changed to include additional graft sources from places such as umbilical cord blood and mobilized peripheral blood stem cells collected by apheresis. Each type of hematopoietic stem cell graft requires a different type of processing and the grafts are often highly manipulated. To reduce the risk of graft-versus host disease, T cells are often depleted or CD34+ cells are isolated and transplanted. HSC transplantation now includes the administration of cells in addition to HSCs. The administration of lymphocytes or T cells from allogeneic donors following transplantation is often used to treat relapsed leukemia. These donor lymphocyte infusions, or DLIs, are also given to speed immune reconstitution and prevent leukemia or cancer relapse. In some cases, leukocytes that have been selected and stimulated are used for donor lymphocyte infusions [1, 2].
The use of cellular therapies to treat cancer is also growing rapidly. Immune cells, isolated and expanded from surgically resected tumors, are an effective cancer adoptive immune therapeutic tool. The administration of these tumor infiltrating leukocytes, or TIL cells, to patients with metastatic melanoma results in an objective clinical response rate of 31% to 35% [3]. When TIL cell therapy for melanoma is combined with myelosuppressive chemotherapy (with stem cell rescue using autologous CD34+ cells isolated from G-CSF-mobilized PBSC products), the response rates increase to 49% to 72% [4]. Autologous NK cells are also being used to treat leukemia, lymphoma, and cancer [5-8]. In some cases NK cells are expanded more than 100-fold before they are administered to patients [7]. In addition to the administration of these immune cells, dendritic cells (DCs) as profession and highly effective antigen presenting cells are being used for several types of cancers vaccines after in vitro pulsing. Both immature and mature DCs are being used.
The improved clinical outcomes associated with cellular therapies has lead to broader utilization and as are result, quality testing is becoming an increasingly important part of cellular therapy. Most clinical cellular therapy products are initially developed and tested in clinical trials at one center, but as soon as a therapy is found to be effective at a single institution, it is tested at other centers and eventually, in multicenter clinical trials. In order for cellular therapies to be exported from one center to another for further clinical evaluation, it's critical to have mechanisms in place to ensure that the cell collection and processing procedures yield safe, effective, and comparable products at all centers. Products manufactured at all centers should also be of similar composition, purity, and potency.
QUALITY TESTING OF CELLULAR THERAPIES
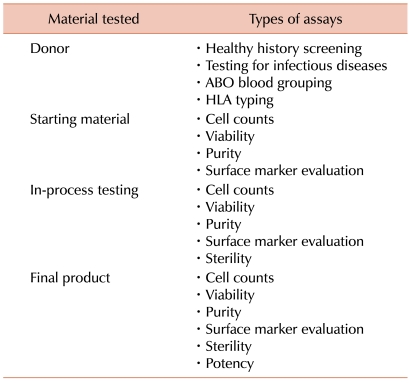
Cellular therapies should be tested at several points during their production (Table 1) thus, the starting material, intermediate products, and the final product are usually all analyzed. Products are evaluated at critical steps in the manufacturing process (in-process testing) and at the end of production prior to the release of the product for clinical use (lot release testing). The results of in-process and lot release assays should fall within specified ranges and meet predetermined acceptance criteria before the product can be released for clinical use. The starting material and final product are generally more thoroughly evaluated than the intermediate products. In addition, the donor of the starting biologic material is usually evaluated as well.
1. Donor testing
The potential donor's medical history is generally assessed. Subjects who have a history of an infectious disease that maybe transmitted by the transfusion of blood or cellular therapy, or have been exposed to such an agent, are often prevented from donating. The donor of the starting material is also generally tested at molecular, antigen and antibody levels for possible exposure to infectious agents that could be transmitted to the cell therapy recipient by the product. The donor's cells, that are used to manufacture products for allogeneic use, for HIV, hepatitis B virus (HBV), and hepatitis C virus (HCV) and may be tested for other blood borne pathogens. The specific pathogens evaluated and the assays used vary between countries and sometimes even within countries. It is usually less critical to test people who are donating cells for themselves. However, autologous donors are often tested for HIV, HBV, and HCV since special precautions may be necessary to protect other patients' cellular therapies and the laboratory staff when processing and storing products from infected donors.
2. Testing the starting material
The cellular starting material is typically tested for volume and cell concentration. Some sort of documentation is usually made, indicating that a sufficient quantity of the desired cell type has been collected. The assessment of the cell type collected may then involve evaluations based on cell size, shape, and morphology or the evaluation of cell surface markers by flow cytometry. The purity of the cells in the starting material is also often evaluated by flow cytometry. In addition, cell viability is also generally measured by dye exclusion assays.
Identity testing is usually performed on the starting material as well. Identity testing measures genetic markers that can be used to confirm that the product's distinctive character which can be used to identify the specific cells at any point in the production process. This is usually achieved by testing for HLA antigens or by ABO blood grouping.
In some cases the starting material is tested for sterility. Sterility testing may include a gram stain, endotoxin testing, and microbial cultures. The starting material may also be tested for mycoplasma using culture or PCR assays.
3. In process testing
During the intermediate processing steps, the product may be tested for volume, cell concentration, cell number, purity, and sterility. It may also be important to document the specific cell populations (such as leukemia or lymphoma cells) that are no longer present. The cells are often characterized by flow cytometry.
4. Final product testing
The testing of the final product generally includes measuring the quantity and purity of the cells. But if the cells have been cultured or extensively manipulated, they should also be tested for sterility.
In addition to the tests discussed above, another important aspect of evaluating the final product is potency testing. Potency testing involves the quantitative measure of biological activity of a product. The biological activity should describe the ability of a product to achieve a defined biological effect. The biological activity measured should be closely related to the product's intended biological effect and ideally, it should be related to the product's clinical response [9-11]. Measurement of the potency of a product is not the same as measuring clinical efficacy. Rather, it is a means to control product quality.
Potency assays for cellular products usually take a considerable amount of time to develop. Generally, the development of potency assays is progressive and begins during preclinical and early clinical development. A potency assay should be validated and already in place prior to phase III clinical trials involving a specific cellular therapy [9, 10].
5. Other quality measures
3 other important measures of cell therapy quality are stability, consistency, and comparability testing. These measures are not performed on all products but are still an important part of product development and ongoing product evaluation.
1) Stability testing
Stability testing is performed to ensure that products remain potent at the time of administration. Since the life of most cells is relatively short (sometimes only hours), stability testing must document cells that are still viable and potent at the end of the several hours required to release the product from the cell therapy laboratory's inventory, transport the product from the laboratory to the patient's bedside, and administer the product to the recipient.
2) Consistency testing
Using the same protocol, there may still be considerable lot-to-lot variability in clinical cellular therapies produced. Consistency testing, therefore, measures variability among products. This variability may be due to differences in starting material or differences in the manufacturing process. Differences in the starting material may be due to variability in the collection process. There may also be differences in the starting material as a result of variability among donors with genetic differences. Genetic polymorphisms in cytokines, growth factors, and their receptors affect the cellular immune response [12-15]. It is likely that these polymorphisms affect the response of cells to cytokine and growth factor stimulation in vitro and the behavior of cells during culture. Epigenetic changes may also be important. The same type of cells obtained from different donors, time points, and physiological conditions could vary significantly due to genetic heterogeneities, epigenetic differences, or transcription regulation diversities. When material is collected from patients for autologous use, there may also be differences due to the patients' disease stage or type, or due to previous therapies.
Variability in the product could also be introduced in the manufacturing process due to differences in equipment, supplies, reagents, and the techniques used by the laboratory staff. Variability due to such production factors can be assessed by measuring the consistency of multiple products manufactured from the same donor or patient. Differences due to donor factors can be assessed by comparing the differences among multiple products manufactured from each donor. Environmentally caused differences can be measured by comparing multiple products manufactured from the same donor or patient at different time points or in different batches.
3) Comparability
It is often necessary to compare products manufactured using different methods. During the scale up of the initial production of a cellular therapy, it is necessary to demonstrate that cells produced on a small scale are the same as those produced with a modified method used for manufacturing cells on a much larger scale. The scaling up of methods often requires the use of different containers, reagents, instruments, and media. These changes could affect the potency of the final product.
Once a specific cellular therapy is being manufactured for patient use, it may be necessary to change the manufacturing process or manufacturing facility due to a change or lack of availability of a reagent, device, or instrument used in the manufacturing process. Cells manufactured using a revised method should be compared with those produced using the original method to ensure that the 2 methods yield comparable cells.
6. Need for molecular testing
The testing of cellular therapies for stability, consistency, comparability and potency is challenging and their evaluation requires the measurement of important functions of the cellular therapies. The function of many cellular therapies is very complex and multiple functions may be required for a therapy to be effective such as trafficking, antigen presentation, cytokine and growth factor release, differentiation, maturation, in vivo expansion, and cytotoxicity. Dendritic cells must traffic to lymph nodes, present antigens, and stimulate T cells. Hematopoietic stem cells must home to their niche, proliferate, and differentiate. For most cell therapies, their critical functions are not completely understood and for some cell therapies, the critical functions are not known. Those unknown functions can only be understood by the application of high throughput technologies which can uncover the entire cell transcriptome and shed light on what the cell intends to do. We have applied global gene and microRNA (miR) expression analysis to cell therapy products and have found that they are very useful for comparability testing and will likely be useful for potency, stability and consistency testing.
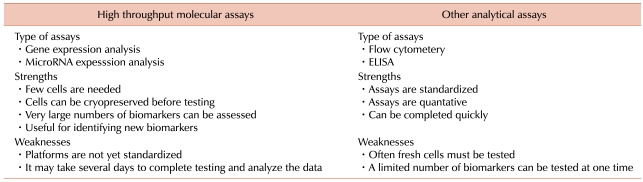
There are several benefits to using gene and miR expression profiling for assessing cellular therapies (Table 2). First, since most cellular therapies are patient-specific, there is usually a limited quantity of suitable source material and therefore, a limited amount of final product available for testing. The starting materials for most cellular therapies are cells collected from human subjects. Generally, an entire production lot of a cellular therapy is usually administered to a single patient and the use of large quantities of the product for testing may adversely affect the dose and clinical effectiveness of the product. This limitation on the quantity of material available prevents the use of some assays and/or limits the number of factors that can be tested.
A practical advantage of gene expression microarrays over other analytical assays such as ELISA or flow cytometry is that it requires very few cells. Enough RNA can be isolated from 1×104 to 1×106 cells for analysis with a global gene cDNA expression microarray [16]. Microarrays with 15,000 to 40,000 genes or oligonucleotide probes have been used clinically to characterize lymphomas, [17] prostate cancer, [18] ovarian cancer, [19] small cell lung cancer, [20] melanoma, [21] and many other cancers. Since gene expression microarrays simultaneously measure the expression of thousands of genes, they capture a snap shot of all possible gene expression signatures that are associated with cellular function and hence, could be a very important tool for assessing the potency of cellular therapies. The comprehensive nature of gene expression microarray analysis makes them ideal for measuring both expected and unexpected cell functions. This is particularly important for the analysis of cells with complex and multiple critical functions such as dendritic cells (DCs), cytotoxic T cells, embryonic stem cells, HSCs, and bone marrow stromal cells (BMSCs).
MicroRNA expression arrays have also been used to access cellular therapies. MicroRNAs are an abundant class of endogenous non-protein-coding small RNAs of 19 to 23 nucleotides that are derived from pre-miRNA of 60 to 120 nucleotides. Mature miRNAs negatively regulate gene expression at the post transcriptional level. They reduce the levels of targeted transcripts as well as inhibit protein translation. More than 800 human miRNAs have been identified so far [22]. In general, miRNAs are phylogenetically conserved and therefore, have conserved and defined post transcription inhibition functions. Some miRNAs are expressed throughout an organism but most are developmentally expressed or are tissue-specific.
MicroRNAs also play an important role in many cellular development and metabolic processes including developmental timing, signal transduction, tissue differentiation, and cell maintenance. Most miRNAs are tissue specific. For example the expression of miR-1 is restricted to the heart [23] while miR-223 is limited to granulocytes and macrophages [24]. Recently, miRNA have been found to play a role in stem cell self-renewal and differentiation. Several different miRNAs are involved with the differentiation of hematopoietic progenitor cells. miR-155, for instance, is important in preventing the differentiation of CD34+ cells toward myeloid and erythroid cells [25] and in the maturation of DCs [26]. In addition, miR-221 and miR-222 prevent the differentiation of hematopoietic stem cells into erythroid progenitors [27] while miR-181 is involved in the control of lymphopoiesis [24].
There are however some limitations concerning the use of gene and miR expression microarrays for potency testing. These platforms are still evolving and it is not always possible to compare the results obtained with different platforms. Gene and miR expression microarray analysis involves multiple steps including RNA isolation, amplification, fluorescent labeling, hybridization, and data analysis. It is impossible with the current technology to complete the whole procedure within a few hours and thus global expression microarrays cannot yet be used for lot release testing. However, if global microarrays can identify specific sets of gene or miR whose expression is associated with potency, tailored chips or quantitative real-time PCR kits that only assess specific "potency genes" could be developed and used for lot release testing.
Despite these limitations, we and many others have found these assays to be very useful in assessing cellular therapies. We present several examples of the use of gene expression analysis for assessing cellular therapies.
EXAMPLES OF THE USEFULNESS OF MOLECULAR ASSAYS
1. In process testing
1) Predicting the confluence of human embryonic kidney 293 cells
Gene expression microarrays can be utilized to predict the quality of cells used to manufacture biological products. Human embryonic kidney (HEK) 293 cells are often used to manufacture products such as adenoviral gene therapy vectors and vaccines [28]. These cells can be grown in bioreactors, tissue culture flasks, and roller bottles. However, when HEK 293 cells have grown to form a confluent monolayer, their phenotype changes as does the quality of the vector or vaccine produced by these cells. Cell confluence can be readily assessed by visual inspection of cells grown in flasks and roller bottles, but for cells grown in bioreactors, the assessment of confluence by visual inspection is not always possible or clear. Gene expression profiling can be used to identify genes whose expression predicts cell confluence [28]. HEK 293 cells that have been grown to 90% confluency have a unique gene expression signature compared to those grown to 40% confluency and therefore a set of 37 of these signature genes is able to predict that quality and confluence of HEK 293 cells.
2) Analysis of embryonic stem cell differentiation
Human embryonic stem cells (hESCs) have the potential to be useful for a number of clinical applications. Since cultured hESCs may undergo spontaneous differentiation, it is important to determine if cultured hESCs have maintained their stem cell qualities or if they have begun to acquire properties of more differentiated cells. Gene expression profiling may thus be useful for assessing cultured hESCs since it has been used to identify genes that are uniquely expressed by hESCs [29]. Player et al have found that 1715 genes were differentially expressed between hESCs and differentiated embryonic cells [29]. This finding is likely to be useful in determining if cells in culture have maintained their embryonic stem cell characteristics.
Embryonic stem cells must be differentiated before they can be used clinically. One of the first steps in the differentiation of hESCs into mature cells and tissues for clinical use is the production of embryoid bodies (hEBs), which involves the aggregation of embryonic cells but the prevention of the differentiation of cells into germ lines by plating them onto a non-permissive substrate. After these hEBs are isolated, they can be induced to generate several different types of cells including hematopoietic cells, neuronal, myogenic, and cardiac muscle cells. A comparison of gene expression profiles of hESCs and hEBs has found 194 genes whose expression was more than 3-fold greater in hEBs than in hESCs [30]. This unique set of genes should also be useful in assessing hESCs differentiation.
hESCs and hEBs also have unique miR expression profiles. Unique miRNA expression patterns have been identified in undifferentiated hESCs. A comparison of miRNA expression profiles among samples from hESCs and hEBs and five different adult cell types revealed that 104 miRNAs differentially expressed miRNA [31]. Seven miRs within miR-302 cluster on chromosome 4 and 21 miRs within the miR-520 cluster on chromosome 19 were highly expressed in undifferentiated hESCs compared to hEBs and the adult cells. The members of these 2 clusters share a consensus 7-mer seed sequence and their targeted genes had overlapping functions. Therefore, miR in the miR-302 and miR-520 clusters are likely to be biomarkers for hESC differentiation. Interest in reprograming adult cells to function back to pluripotent stem cells, induced pluripotent stem (IPS) cells, for regenerative medicine applications is growing and gene and miR expression analysis will also likely be effective in assessing these cells.
2. Comparability testing
We have also found gene expression analysis to be useful for comparability testing. By applying gene expression analysis we have shown that immature DCs made from fresh PBMCs are a similar transcription profile to those made from stored PBMCs [32] and that mDCs produced with three different maturation protocols share similar transcriptome profiles [33]. The conclusion that two types of immature DCs and three types of mDC were similar could only be made when genome wide whole transcripts were analyzed. This suggests that gene expression analysis is useful for comparability testing.
1) Immature DCs from stored PBMCs
The production of clinical cellular therapies often involves multiple centers. Cells such as PBMCs are collected from a donor or patient at one center and is then shipped to a specialized cellular therapy laboratory where they are processed and become DCs. When production of the cellular therapy at the centralized center is complete, the product is shipped to the site where it is administered to a specific patient. But the centralized cell processing laboratory may be hundreds or thousands of miles from the collection center and it may even take 48 hours or more after the collection is complete for the starting cellular material to reach the cell processing laboratory.
To evaluate whether immature DCs (iDCs) produced from fresh PBMCs are similar to those produced from PBMCs that have been stored for 48 hours at 4℃ and treated with GM-CSF and IL-4, we evaluated gene expression profiles of both types of iDCs [32]. Global gene expression analysis revealed that 48 hours of storage at 4℃ had some effects on the PBMCs, but had no effect on the iDCs. The striking similarity in gene expression among iDCs derived from fresh and stored PBMCs suggests that there is no difference in the function of these cells and that the changes in PBMCs during storage did not effect the ability of these cells to be used to produce iDCs.
2) Comparison of DC maturation protocols
Some adoptive immune therapy studies have used iDCs, however, mature DCs (mDCs) are increasingly being used for clinical immune therapy because they express increased levels of co-stimulatory molecules, produce greater quantities of cytokines, and are superior in the stimulation of cytotoxic T-cell response compared to iDCs [34]. mDCs with effective anti-tumor responses have high levels of expression of co-stimulatory molecules CD80, CD83, and CD86, have high migratory activity in response to the lymphoid organ chemokines CCL19 and CCL21, [35, 36] and have high capacity to produce interleukin-12p70 (IL-12p70) which is the major factor driving Th1 reactions [37, 38].
To produce mDCs, imDCs are treated with various stimuli known to induce DC maturation [37, 39, 40]. DC maturation can be induced using inflammatory signals such as tumor necrosis factor (TNF), interleukin-1beta (IL-1β), or bacterial derivatives such as lipopolysaccharide (LPS). Double stranded RNA, interferons and postaglandins can also induce DC maturation [41, 42]. The "gold standard" DC maturation cocktail has been IL-1β, IL-6, TNF-α, and prostaglandin E2 (PGE2) [43]. This cocktail generates mDCs with high co-stimulatory and migratory functions, however they produce relatively low levels of IL-12 [44]. As a result, a number of ex vivo maturation cocktails have been developed for clinical use that generate mDC that produce higher levels of IL-12p70 than the IL-1β, IL-6, TNF-α and PGE2 cocktail [37].
LPS is a toll-like receptor (TLR) ligand and a microbial compound originating from the outer membrane of the gram-negative bacterial cell wall. It is being used as a DC maturation stimulant since it is a good inducer of IL-12p70 production [38, 40]. LPS stimulates Toll-like receptor 4 (TLR4), mediates the activation of nuclear factor-kappa B (NF-kB) and mitogen activated protein kinases (MAPKs), and finally initiates DC maturation [45, 46]. Another important DC maturation stimulant is IFN-γ which enhances the TLR signaling pathway and up-regulates DC inflammatory cytokines. It has been also reported that IFN-γ can significantly augment IL-12p70 production [47].
In order to determine if the maturation of DCs by the combination of LPS plus IFN-γ can be improved by the addition of IL-1β or IL-1β plus TNF-α, we compared mDCs produced from PBMCs with these agents. Monocyte-derived DCs were prepared by the elutriation of monocytes from PBMCs followed by incubation in GM-CSF and IL-4 to produce iDCs. To produce mDCs, the iDCs were stimulated with LPS plus IFN-γ; LPS, IFN-γ, plus IL-1b; and LPS, IFN-γ, IL-1β plus TNF-α. All three DC maturation cocktails produced mDCs that expressed greater levels of commonly used markers of DC maturation: CD80, CD83, CD86, and CCR7. In addition, there were no differences in the production of IL-12p70 and IL-10 by the mDCs produced with the 3 different cocktails.
Although traditional assays used to assess DCs, flow cytometry and ELISA, did not reveal any differences between the three groups of mDCs, the function of DCs is complex and still not completely understood. It is likely that DC function is influenced by many other factors in addition to the expression of CD80, CD83, CD86, and CCR7 and the production of IL-12p70 and IL-10. To more thoroughly compare the mDCs produced with the three maturation cocktails, we analyzed the mDCs using gene expression microarrays with more than 36,000 probes. While we found that 9,576 genes were differentially expressed between iDCs and mDCs, the expression of only 13 genes differed between the three groups of mDCs. Multivariate analysis also suggested that there were more differences between DCs from different donors than between maturation cocktails [33]. These results thus show that there is no benefit of adding IL-1β and TNF-α to the LPS plus IFN-γ DC maturation cocktail.
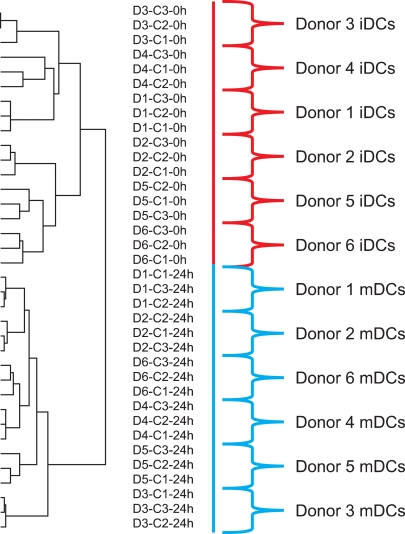
3. Consistency testing
Global gene expression analysis may be an excellent tool for assessing the consistency of cellular therapies. In the study described above, in which we evaluated mDCs produced with three different maturation cocktails (LPS plus IFN-γ; LPS, IFN-γ plus IL-1β; and LPS, IFN-γ, IL-1β plus TNF-α) from six healthy subjects, the DCs clustered according to donor rather than maturation protocol (Fig. 1). This result suggests that global gene expression profiling can distinguish differences due to donor variability. The ability of gene expression profiling to detect minor differences between healthy subjects will likely make this an excellent assay for consistency testing.
A number of cellular therapy production protocols involve the serial production and administration of cellular therapies produced from autologous cells. Global gene expression analysis should allow for the assessment of differences among multiple products produced from the same donor that may be due to problems or modifications in the cell therapy production procedure.
4. Potency testing
1) Potency testing of hematopoietic stem cells
Potency assays used for HSC products should measure the ability of the product to reconstitute bone marrow hematopoietic cells and peripheral blood cells in the transplant recipient. The potency assay should also reflect the period of time that neutrophil, platelet, and red blood cell counts return to and remain above specified levels independent of transfusion therapy.
Liquid culture of long-term culture initiating cells (LTC-IC) and the repopulation of marrow in nonobese diabetic (NOD)/severe combined immunodeficiency (SCID) mice assays are considered to be the best measure of the quantity and quality of hematopoietic stem cells. However, these assays require highly specialized reagents, a highly trained staff, and several weeks to complete. As a result, these assays have seldom, if ever, been used as potency assays. The measurement of myeloid, erythroid, and mixed colony formation in methylcellulose culture systems has been used for assessing bone marrow and PBSC HSC products, however, these assays take approximately 14 days to complete and do not produce reliable quantitative data. Traditionally, total nucleated cell counts were used to assess the potency of bone marrow and are still used as a measure of potency of umbilical cord blood components prepared for transplantation. The measurement of CD34+ cells by flow cytometry has, however, become the universal assay for measuring the potency of HSC products. One limitation of the use of CD34+ antigen expression as a potency marker is that HSCs expressing CD34 antigen do not represent a homogenous population. Several distinct subpopulations or phenotypes of CD34+ cells have been described [48]. Some subpopulations are more primitive while others are more likely to differentiate into myeloid cells, erythroid cells or megakaryocytes.
Recently, the agent plerixafor has been approved by the US Food and Drug Administration for use in combination with G-CSF to mobilize stem cells for autologous transplantation. The mechanisms by which plerixafor and G-CSF alter HSC trafficking and mobilization are different, suggesting that HSCs with different intrinsic properties may be mobilized by these agents. Plerixafor, as a CXCR4 antagonist, mobilizes HSCs within 6 hours [49-51] by disrupting the engagement of stem cell surface CXCR4 [49, 52, 53] with its ligand SDF-1 (CXCL12) which is expressed on marrow osteoblast [54, 55]. In contrast, G-CSF mobilizes stem cells indirectly by down-regulating the expression of CXCL12 on marrow osteoblasts and by releasing neutrophil and monocyte proteolytic enzymes including neutrophil elastase, cathepsin G, and matrix metalloproteinase-9 which in turn degrade important HSC trafficking and adhesion molecules c-kit, VCAM-1 and CXCR4 [56].
To explore the possibility of using gene and miRNA expression profiles assess HSC potency, we used a rhesus macaque HSC-mobilization model to compare CD34+ cells mobilized by G-CSF, by plerixafor, and by the combination of G-CSF and plerixafor. The CD34+ cells were compared using global gene and miR expression analysis. While many of the same genes were up-regulated in CD34+ cells mobilized by either plerixafor or G-CSF, there were also a large number of genes expressed that differed [57]. G-CSF-mobilized CD34+ cells were more likely to express neutrophil and mononuclear phagocyte genes than those mobilized by plerixafor. In addition, G-CSF-mobilized CD34+ cells also expressed high levels of miR-155, which is expressed in maturing DCs [26, 58]. In contrast, plerixafor-mobilized CD34+ cells were more likely to express B, T, and mast cell genes. These results suggest that G-CSF mobilizes a greater proportion of HSCs that are committed toward myeloid differentiation while those mobilized by plerixafor are more likely to be committed toward B, T, and mast cells.
Surprisingly, the combination of plerixafor plus G-CSF mobilized a population of CD34+ cells that were distinct from CD34+ cells mobilized by either agent alone. CD34+ cells mobilized by plerixafor plus G-CSF were more likely to express B cell and T cell genes than G-CSF-mobilized and plerixafor-mobilized CD34+ cells. In addition, plerixafor plus G-CSF-mobilized CD34+ cells were also remarkable for their increased expression of miR142-3p and miR-142-5p. miR-142-3p and miR-142-5p are highly expressed in T cells [59] and miR-142-3p has been found to be increased in childhood B-cell precursor acute lymphoblastic leukemia [60]. These results thus suggest plerixafor plus G-CSF mobilizes a greater proportion of B and T cell precursors than either G-CSF or plerixafor alone.
Other studies have also found that plerixafor, G-CSF, and plerixafor plus G-CSF mobilize different types of CD34+ cells. In a mouse model, Pitchford and colleagues have found that G-CSF mobilized greater quantities of hematopoietic progenitor cells than plerixafor while plerixafor mobilized greater quantities of endothelial progenitor cells than G-CSF [61]. They also found that the combination of G-CSF plus plerixafor mobilized greater quantities of hematopoietic progenitors than either agent alone, but not endothelial progenitors. Additionally, Jin et al have found that HSCs in healthy subjects mobilized with plerixafor or plerixafor plus G-CSF differ from those mobilized by G-CSF alone [62]. Furthermore, flow cytometry analysis by Rettig and colleagues of CD34+ cells mobilized by Plerixafor alone identified a unique CD34dimCD45RA+ subset that was not mobilized by G-CSF alone [63]. The results of these studies demonstrate a role for gene and miR expression analysis for HSC potency testing.
CONCLUSIONS
As more and more new cellular therapies are being developed and used to treat an increasing variety of diseases and patients, the analysis of the quality of cellular therapies (including stability, consistency, comparability, and potency testing) is becoming a critical and required part of their production. Existing assays such as function, flow cytometry, and ELISA are important, but gene and miRNA expression microarrays have the potential to become essential additions to the standard assays for assessing cellular therapies. Gene and miR assays have already been shown to be useful in comparability and stability testing. These assays are also well suited for assessment of the potency of cellular therapies in phase I and II clinical trials. During these early phase clinical trials, genes and miRs whose expression is found to be associated with critical biological function can be identified and evaluated as possible biomarkers of potency.
 XML Download
XML Download