

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The prognoses of high-risk medulloblastoma (MB) and supratentorial primitive neuroectodermal tumor (sPNET) are poor despite the combined use of chemotherapy and radiotherapy [1, 2]. Although the 5-year event-free survival (EFS) rate for patients with average-risk MB or sPNET is 60-80% [3-5], the 5-year EFS rate for patients with high-risk MB or sPNET is 30-60% depending on the treatment received [1, 2]. Moreover, in cases with proven metastasis, the prognoses are worse. To improve the outcome of high-risk MB and sPNET, two obstacles that greatly affect the quality of life for patients, treatment-related mortality (TRM) and long-term sequelae, must be obviated. The currently available treatments for patients with high-risk MB or sPNET are plagued by considerable treatment-related toxicity due to the increased vulnerability of the central nervous system in young patients and radiation therapy-related significant long-term neurological sequelae [6-8]. All patients diagnosed with MB conventionally receive 36 Gy of craniospinal radiation therapy (CSRT) with a boost of 18-20 Gy to the posterior fossa. Recently, reduced-dose CSRT with 23.4 Gy and a boost to the posterior fossa have been used for average-risk MB; however, 36 Gy of CSRT is still used for high-risk patients [5, 9, 10]. The fact that survivors of MB who received 23.4 Gy instead of 36 Gy of CSRT experienced less severe neuropsychological toxicity prompted us to develop a strategy involving reduced-dose CSRT and intensified tandem high-dose chemotherapy with autologous stem cell rescue (HDCT/ASCR) [11]. Recently, we examined the efficacy and safety of tandem HDCT/ASCR for high-risk neuroblastoma based on the theory that dose escalation may improve survival, and we found improved survival rates [12, 13].
Strother et al. [14] proposed HDCT/ASCR for the treatment of MB and sPNET, and their progression-free survival rates suggested that HDCT/ASCR is a feasible option for patients with average- and high-risk MB or sPNET. However, data showing the efficacy and long-term sequelae of HDCT/ASCR in children with high-risk brain tumors are limited.
Thus, in this study, we prospectively investigated the effects of reduced-dose CSRT followed by tandem HDCT/ASCR in children with a newly diagnosed high-risk MB or sPNET through a multicenter study with a single-arm regimen in an effort to reduce the incidence of long-term sequelae and improve survival.
MATERIALS AND METHODS
1. Patients
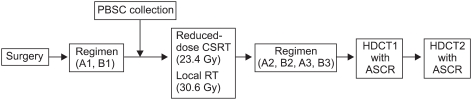
Between March 2005 and April 2007, newly diagnosed high-risk MB and sPNET patients older than 3 years received treatment according to the Korean Society of Pediatric Neuro-Oncology (KSPNO) protocol S051 for high-risk brain tumors (Fig. 1) at the Samsung Medical Center, Asan Medical Center, Seoul National University Hospital, and Hanyang University Hospital, in Seoul, Korea. The patients were defined as high-risk if they had metastatic disease at presentation (M+) or a residual tumor exceeding 1.5 cm2 after surgery.
This study was approved by the institutional review board at each participating hospital. Written informed consent was obtained from the parents of each patient.
2. Pre-HDCT chemotherapy and radiotherapy
Initially, maximum resection of the tumor was attempted by a pediatric neurosurgeon at the participating centers. We defined the extent of resection using the neurosurgeons' operative notes and magnetic resonance images (MRIs) obtained after surgery.
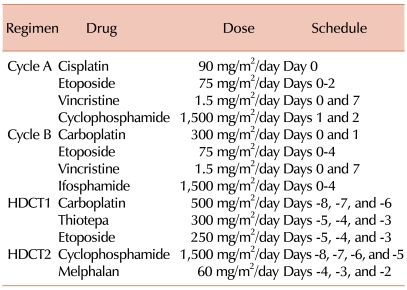
The patients were scheduled to receive 2 cycles of pre-RT chemotherapy within 4 weeks after surgery. Cycle A consisted of cisplatin (90 mg/m2/day on day 0), etoposide (75 mg/m2/day on days 0-2), vincristine (1.5 mg/m2/day on days 0 and 7), and cyclophosphamide (1,500 mg/m2/day on days 1 and 2); cycle B consisted of carboplatin (300 mg/m2/day on days 0 and 1), etoposide (75 mg/m2/day on days 0-4), vincristine (1.5 mg/m2/day on days 0 and 7), and ifosphamide (1,500 mg/m2/day on days 0-4) (Table 1). Radiotherapy consisted of 23.4 Gy (1.8 Gy per fraction, 5 fractions per week) to the craniospinal axis, with 30.6 Gy to the primary tumor. In cases with M2 or M3 stage disease, an additional 21.6 Gy was applied to the seeding nodule. Within 4 weeks after RT, post-RT chemotherapy was initiated when the absolute neutrophil count exceeded 1,000/µL and the platelet count exceeded 75,000/µL. The patients were scheduled to receive cycles A and B alternating at 4-week intervals for 4 cycles (cycles A2, B2, A3, and B3). The doses used for post-RT chemotherapy were reduced to 75% of the pre-RT values, except for vincristine. After chemotherapeutic cycle B2, if the hematopoietic recovery was delayed for more than 2 weeks, the doses used in cycles A3 and B3 were reduced to 50% of their pre-RT values, except for vincristine. After four cycles of post-RT chemotherapy, tandem HDCT/ASCR was performed (Fig. 1).
3. Collection of peripheral blood stem cells (PBSCs)
Hematopoietic stem cells were collected from granulocyte-colony-stimulating factor-mobilized peripheral blood after cycle A1. We attempted to collect at least 2.0×106 CD34+ cells/kg for tandem HDCT, and at least 1.0×106 CD34+ cells/kg for each round of HDCT. If the collected PBSCs were not sufficient for tandem HDCT, we attempted to collect PBSCs again after cycle B1.
4. HDCT with ASCR
For the first round of HDCT (HDCT1), CTE (500 mg/m2/day carboplatin on days -8, -7, and -6; 300 mg/m2/day thiotepa on days -5, -4, and -3; and 250 mg/m2/day etoposide on days -5, -4, and -3) was used. Twelve weeks after day 1 of HDCT1 and after recovery from organ failure, CM (1,500 mg/m2/day cyclophosphamide on days -8, -7, -6, and -5, and 60 mg/m2/day melphalan on days -4, -3, and -2) was given to the patients as a conditioning regimen for the second round of HDCT (HDCT2) (Table 1 and Fig. 1). After HDCT, autologous stem cells were infused on day 0 for marrow rescue.
5. Response and toxicity criteria
The responses of the patients were categorized as follows: progressive disease (PD), >25% increase in tumor size or the appearance of a new tumor; stable disease (SD), <25% decrease or increase in tumor size; minor response (MR), 25-50% decrease in tumor size; partial response (PR), >50% decrease in tumor size; or a complete response (CR), complete disappearance of all previously measurable tumors [15]. Relapse was defined as a lesion that developed at a new site (i.e., a site that was previously tumor free). MRIs were used to evaluate the disease status of all patients. Adverse events were evaluated according to the National Cancer Institute's Common Toxicity Criteria (NCI-CTC).
6. Statistical analysis
EFS was estimated from the date of the primary surgery to the date of relapse or disease progression or to the last follow-up date for those patients who did not experience any of these events. Overall survival (OS) was measured from the date of the primary surgery to the date of death for any cause or of the last follow-up. Kaplan-Meier estimates of the survival distribution were calculated and compared using the log-rank test. Variables were compared between groups using the chi-square test. Statistical significance was accepted for P-values<0.05.
RESULTS
1. Patient characteristics
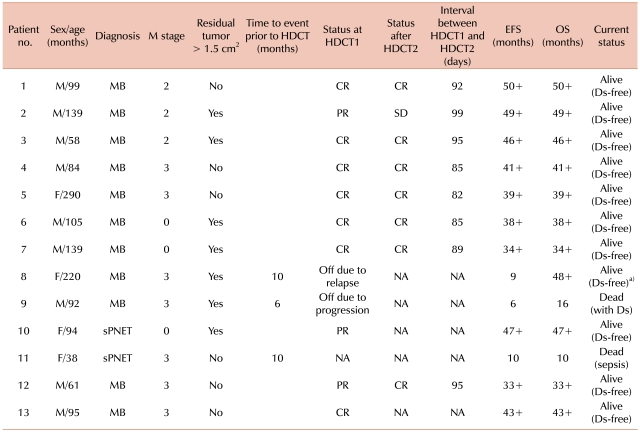
Thirteen patients (9 boys and 4 girls) older than 3 years with a newly diagnosed MB (n=11) or sPNET (n=2) were enrolled. The median age at diagnosis was 95 months (range 38-290). Seven patients (53.8%) had a residual tumor >1.5 cm2 after surgery. Ten patients (76.9%) had evidence of metastatic disease at diagnosis: 3 were stage M2, and 7 were stage M3 (Table 2).
2. Response and outcome of HDCT with ASCR
Of the 13 patients, 1 (No. 9) died from PD and another (No. 11) died from septic shock after cycle B2. Patient No. 8, who relapsed after cycle B3, received RT followed by HDCT1 and HDCT2 and is currently alive. Ten patients received HDCT: eight received tandem treatment, whereas two patients received HDCT1 only because they refused further treatment. At the time of HDCT1, 7 patients were in CR and 3 in PR. In comparison, at the time of HDCT2, 7 patients were in CR and 1 in SD. The median time from HDCT1 to the initiation of HDCT2 was 90.5 days (range 82-99) (Table 2).
3. Hematologic recovery
The median numbers of total nuclear cells and CD34+ cells/kg infused for HDCT1 were 16.1×108 (range 2.8-20.92) and 36.1×106 (range 8.0-220.2), and for HDCT2 they were 24.2×108 (range 3.1-48.9) and 33.9×106 (range 15.8-157.0), respectively.
Hematologic recovery was achieved rapidly in all patients. The median time to neutrophil recovery was 8 days (range 7-11) for HDCT1 and 9 days (range 8-10) for HDCT2; the median time required to reach a platelet count of 20,000/µL without transfusion for 7 days was 17 days (range 7-54) for HDCT1 and 20 days (range 9-21) for HDCT2.
4. Survival Rates
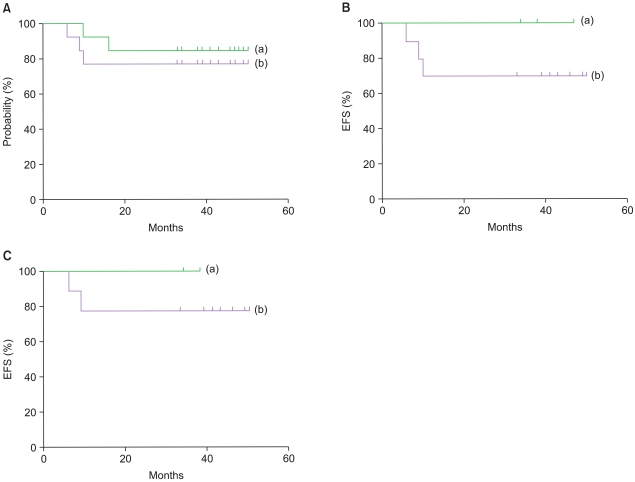
The median follow-up duration was 39 months (range 6-50). The probability of 3-year EFS for the 13 patients was 76.9±11.7%, whereas the 3-year OS was 84.6±10.0% (Fig. 2A). The 3-year EFS rate for those patients who actually received HDCT was 100%. The 3-year EFS rate for the Mo patients was 100%, whereas the rate for the M+ patients was 70.0±14.5% (Fig. 2B). One of the 11 patients was diagnosed with a MB relapsed, and another had PD prior to HDCT; hence, the 2-year EFS of the MB patients was 81.8±11.6%. Among the 9 patients with MB who received HDCT, 8 received tandem double HDCT with ASCR, and none exhibited a relapse or TRM. The 3-year EFS rate for the M0 patients with MB was 100%, whereas the rate for the M+ patients at the time of diagnosis was 77.8±13.9% (Fig. 2C). One of the 2 sPNET patients received HDCT1; however, no further treatment was given based on the wishes of the parents, and another patient died of sepsis after chemotherapy cycle B2.
5. Toxicity
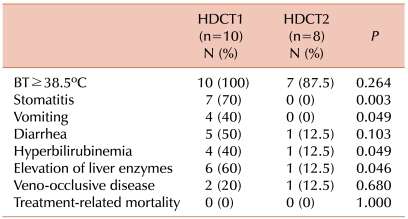
Ten (100%) and 7 (87.5%) patients suffered from a high fever (body temperature ≥38.5℃) during HDCT1 and HDCT2, respectively. The incidence of a grade 3-4 fever, vomiting, diarrhea, veno-occlusive disease (VOD), hyperbilirubinemia, and elevations of liver enzymes during HDCT1 and HDCT2 is shown in Table 3. Of these, the most frequent toxicities were fever, stomatitis, and elevations of liver enzymes during HDCT1. More adverse events occurred during HDCT1 than during HDCT2, but no TRM was observed (Table 3).
DISCUSSION
The clinical outcome for patients with a MB varies according to age, tumor remnants, and metastatic stage [1, 16]; therefore, risk-adapted treatments are required to not only increase survival but to also reduce toxicity that can impact on a patient's quality of life. Among pediatric brain tumors, MB has the greatest potential for extraneural spread. In patients with a metastatic (M2-3) MB, progression-free survival rates of 24-40% have been reported [17-19]. Although combined treatment with chemotherapy and radiotherapy has improved the survival rate in recent years, a substantial number of patients have recurrent PD after multimodal treatment, including surgery, radiation, and chemotherapy [1-3]. Recently, HDCT with ASCR has been used to treat pediatric patients with advanced solid tumors or brain tumors and to avoid the deleterious side-effects of RT in infants and very young children with brain tumors [12, 15, 20]; however, few studies have demonstrated the utility of HDCT with ASCR in the front-line treatment of young children with high-risk MB or sPNET [14, 15, 20].
A 10 Gy dose of CSRT could lead to a 10-point decrease in the intelligence quotient of the recipient and significantly increases the risk of growth hormone deficiency. Packer et al. [21] reported a growth hormone deficiency in all patients who received 36 Gy of CSRT and in 50% of patients who received 23.4 Gy. The extent to which RT could be decreased, in dose or volume, without a substantial increase in the disease relapse rate is unknown; thus, the CSRT dose should be reduced to the smallest dose that does not result in the relapse of the disease, and together with HDCT, could improve the OS and reduce the risk of RT-related cognitive and endocrine defects [20, 21]. Gajjar et al. [20] have shown a special interest in reducing the amount of chemotherapy not only in terms of dosage but also of duration. This phase II clinical trial used risk-adapted CSRT followed by a short, dose-intensive, cyclophosphamide-based regimen with serial, autologous PBSC rescues, resulting in a 5-year EFS rate of 85% in average-risk patients and 70% in high-risk patients.
In our study, 23.4 Gy CSRT followed by tandem HDCT with ASCR was acceptable in terms of toxicity and the survival rate. The 3-year EFS rate for high-risk MB and sPNET patients was 76.9%. One patient had a relapse, another patient progressed before HDCT, and a third patient died of sepsis after chemotherapy; however, the 3-year EFS rate for those patients who actually received HDCT with ASCR was 100%. Compared with other studies of high-risk brain tumors, the EFS rate of our patients was encouraging, and no toxicity-related death occurred during tandem HDCT; however, the optimal therapy for patients with a relapse or PD before HDCT with ASCR should be investigated further. The patients who relapsed or progressed before HDCT with ASCR were older and had stage M3 disease at the time of diagnosis. Therefore, patients older than 6 years of age or with M2-3 disease at presentation may need a higher dose CSRT (at least 30.6 Gy).
Carboplatin, etoposide, and thiotepa were used for HDCT1, whereas cyclophosphamide and melphalan were used for HDCT2 in this study. These regimens were tolerable, and no toxic death or relapse was observed after HDCT. Busulfan was avoided, as it might cause hepatic VOD. In a recent retrospective study, Cheuk et al. [22] speculated that for patients with a MB or sPNET, treatment with carboplatin and thiotepa or etoposide was among the safest and most effective regimens. The optimal combined regimen for consecutive tandem HDCT has yet to be determined for patients with high-risk brain tumors; however, we suggest that the use of different drug combinations between the first and second round of HDCT is a reasonable approach to avoid drug resistance and overlapping drug toxicities [15].
It has been speculated that the second round of therapy in tandem HDCT should be initiated soon after HDCT1 to eradicate possible residual tumor cells and to prevent a relapse, and that delayed HDCT2 might be a cause of relapse [15]. However, in the present study, a high rate of adverse events occurred during HDCT1 compared to HDCT2; thus, sufficient rest was needed after HDCT1. The high rate of adverse events appeared to be related to differences in the intensities of the regimens, but no TRM occurred during tandem HDCT.
In this study, the follow-up durations were too short to evaluate neurological sequelae or hormonal imbalances. The reduced-dose CSRT before HDCT with ASCR may have a favorable impact not only on survival but also on the patient's quality of life, especially in terms of neurological function and hormonal balance. At present, we are conducting outcome studies of neurological and hormonal functions in the long-term survivors of this study.
Although the follow-up period was short and the patient cohort was small in size, the results of this study are encouraging. The limited toxicity and favorable EFS rate observed in children treated with reduced-dose CSRT followed by tandem HDCT and ASCR warrant further exploration in a larger study population.
 XML Download
XML Download