

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Background:
The aim of this study was to investigate the prevalence, clinical and laboratory findings of hereditary hemolytic anemia (HHA) in Korea from 1997 to 2006 and to develop the appropriate diagnostic approach for HHA.
Methods:
By the use of questionnaires, information on the clinical and laboratory findings of HHA diagnosed from 1997 to 2006 in Korea was collected and analyzed retrospectively. A total of 431 cases were enrolled in this study from 46 departments of 35 hospitals.
Results:
The overall frequency of HHA did not change through the 10-year period for pediatrics but did show an increasing tendency for internal medicine. The overall male to female sex ratio did not show sex predominance (1.17:1), but a significant male predominance with a ratio of 1.49:1 was seen for pediatrics while a significant female predominance with a ratio of 1:1.97 was seen forinternal medicine. Of the total cases, 74.2% (282/431) were diagnosed before the age of 15 years. The etiologies of HHA were classified as red cell membrane defects, hemoglobinopathies, red cell enzyme deficiencies and unknown causes. There were 382 cases (88.6%) of red cell membrane defects with 376 cases (87.2%) of hereditary spherocytosis and 6 cases (1.4%) of hereditary elliptocytosis, 20 cases (4.6%) of hemoglobinopathies with 18 cases (4.2%) of β-thalassemia, a case (0.2%) of α-thalassemia and a case (0.2%) of Hemoglobin Madrid, 7 cases (1.6%) of red cell enzyme deficiencies with 5 cases (1.2%) of glucose-6-phosphate dehydrogenase (G-6-PD) deficiency, a case (0.2%) of pyruvate kinase (PK) deficiency and a case (0.2%) of enolase deficiency, and 22 cases (5.1%) of unknown causes. The most common chief complaint in pediatric patients was pallor and that in adult patients was jaundice. In the red cell membrane defect group of patients, the level of hemoglobin was significantly higher than in adult patients. The mean corpuscular volume, mean corpuscular hemoglobin, corrected reticulocyte count, total and indirect bilirubin level and lactate dehydrogenase levels in the hemoglobinopathy group of patients were significantly lower than the values in the red cell membrane defect group of patients. The mean concentration of G-6-PD was 0.8±0.7U/1012RBC in the G-6-PD deficient patients, PK was 1.7U/1010 RBC in the PK deficient patient, and the level of enolase was 0.04U/g of Hb in the enolase deficient patient.
Conclusion:
The most prevalent cause of HHA in Korea during 1997 to 2006 was hereditary spherocytosis, but HHA by other causes such as hemoglobinopathy and red cell enzyme deficiency gradually increased with the development of molecular diagnostic methods and increasing general interest. However, the etiologies of HHA need to be pursued further in 5.1% of the patients. An systematic standard diagnostic approach is needed in a nationwide prospective study for correct diagnoses and appropriate management of HHA.
Go to : 
REFERENCES
1). Hah JO. Hereditary hemolytic anemia. J Korean Med Assoc. 2006. 49:908–19.

2). Ahn DH., Sohn KC., Kang IJ, et al. Statistical analysis of hemolytic anemia in Korea. Korean J Hematol. 1991. 26:445–61.
3). Park JC., Kim YJ., Seo JJ., Moon HN., Ghim T., Park JE. A clinical study on hereditary spherocytosis. Korean J Pediatr Hematol-Oncol. 2000. 7:9–15.
4). Kwon HS., Kang JC., Won SC., Oh SH., Lyu CJ. A clinical study on childhood hemolytic anemia according to etiological classification. J Korean Pediatr Soc. 2003. 46:883–8.
5). Kim DK., Han DK., Baek HJ., Kook H., Hwang TJ., Yeo CY. A clinical study of hereditary spherocytosis. Chonnam Med J. 2006. 42:174–9.
6). Lukens JN. Abnormal hemoglobins: General principles. Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader B, editors. Wintrobe's clinical hematology. 11th ed.Philadelphia, USA: Lippincott Williams & Wilkins Press;2004. p. 1247–62.
7). Koo MS., Lee HS., Ahn YM., Cho HI., Kim JQ., Kim SI. A case of β-thalassemia minor in a Korean family-The first case report in Korea-. Korean J Clin Pathol. 1988. 8:153–60.
8). Park SS., Lee YJ., Kim JY, et al. β-thalassemia in the Korean population. Hemoglobin. 2002. 26:135–45.
9). Lee YJ., Park SS., Kim JY., Cho HI. RFLP haplotypes of β-globin gene complex of β-thalassemic chromosomes in Koreans. J Korean Med Sci. 2002. 17:475–8.
10). Red cell enzymopathies. In: Cha YJ, eds. Hematology. Seoul, Korea: E-public. 2006. 111–6.
11). Lee GB., Lee SJ., Kim YJ, et al. A case of G-6-PD Guadalajara. Korean J Pediatr. 2004. 47:210–3.
12). Park-Hah JO., Kanno H., Kim WD., Fujii H. A novel homozygous mutation of PKLR gene in a pyruvate-kinase-deficient Korean family. Acta Haematol. 2005. 113:208–11.
13). Lee YK., Cho HI., Park SS, et al. Abnormalities of erythrocyte membrane proteins in Korean patients with hereditary spherocytosis. J Korean Med Sci. 2000. 15:284–8.
14). Agre p. Asimos A., Casella JF., McMillan C. Inheri-tance pattern and clinical response to splenectomy as reflection of erythrocyte spectrin deficiency in hereditary spherocytosis. N Engl J Med. 1986. 315:1579–83.
15). Elghetany MT., Banki K. Erythrocytic disorders. In: McPherson RA, Pincus MR, eds. Henry's clinical diagnosis and management by laboratory methods. 21st ed. Philadelphia, USA: Saunders Elsevier. 504–44.
Go to :
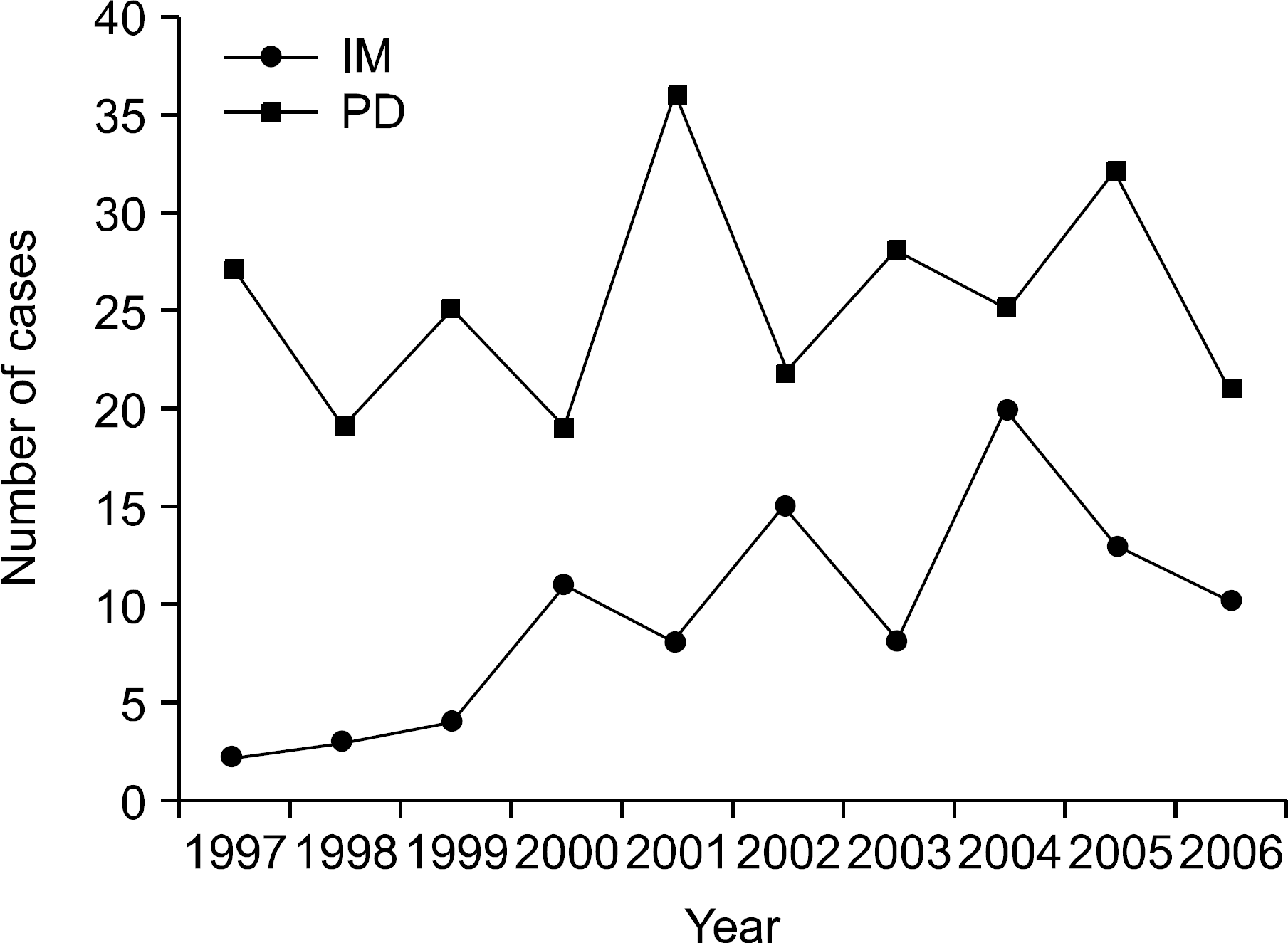 | Fig. 1The annual number of newly diagnosed patients. Abbreviations: IM, internal medicine; PD, pediatrics. |
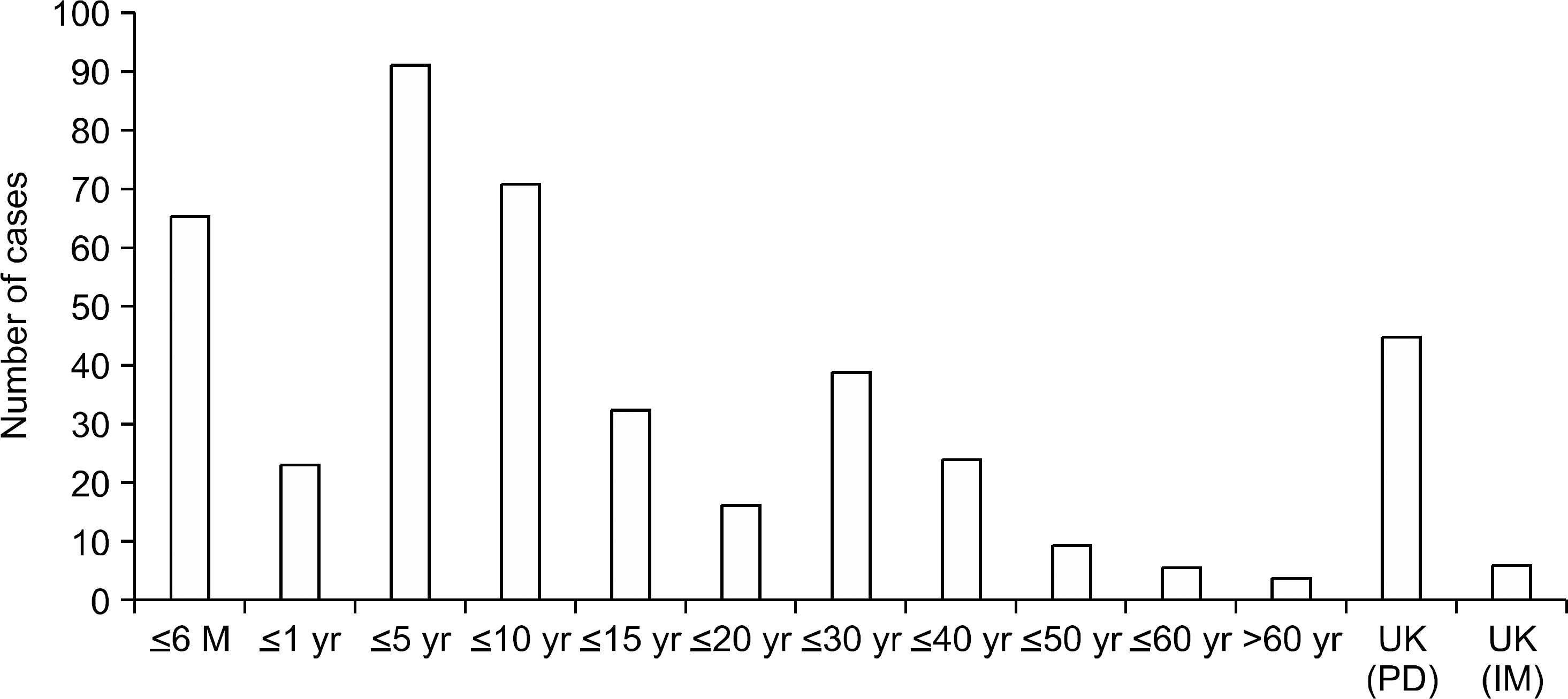 | Fig. 2Age distribution on diagnosis. Abbreviations: M, month; yr, year; UK, unknown; IM, internal medicine; PD, pediatrics. |
Table 1.
The hospitals and departments participating in this study
Table 2.
Sex distributions of patients
| M (%) | F (%) | Total (%) | |
|---|---|---|---|
| IM | 33 (33.7) | 65 (65.3) | 98 (100) |
| PD | 199 (59.8) | 134 (40.2) | 333 (100) |
| Total | 232 (53.8) | 199 (46.2) | 431 (100) |
Table 3.
Etiologies of hereditary hemolytic anemia
Table 4.
Symptoms and signs of hereditary hemolytic anemia
Table 5.
Laboratory findings on diagnosis
Abbreviations: IM, internal medicine; PD, pediatrics; Memb, membrane; Hgb, hemoglobin; Enz def, enzyme deficiency; UC, unclassified; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; RDW, red cell distribution width; WBC white blood cell; C-reti, corrected reticulocyte count; TB, total bilirubin; IB, indirect bilirubin; LDH, lactate dehydrogenase; G-6-PD, glucose-6-phosphate dehydrogenase; HgA, hemoglobin A; HgA2, hemoglobin A2; HgF, hemoglobin F.
 XML Download
XML Download