

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
The human genome contains 3×109 bp of nucleotides and is estimated to have approximately 25000 protein-coding genes. However, humans produce more than 25000 proteins that account for the overall functions of the body. Indeed, the word “proteoform” is used to designate all the different molecular forms in which the protein product can be found, including alterations by genetic variations, alternatively spliced RNA transcripts, and post-translational modifications.1Humans are estimated to have 1×106 “proteoforms”.2 To explain this discrepancy, the biochemical term adopted to describe multifunctional proteins that have a primary well-known function is “moonlighting,” which refers to having a secondary role in addition to the primary role.3 Moonlighting proteins might play a role in bridging the gap between the gene and protein numbers, and a growing number of moonlighting proteins have been identified to play key roles in several human diseases.
Moonlighting proteins possess multiple functions not from gene fusions, splice variants, or protein fragments expressing different functions after proteolysis.4 Currently, approximately 500 moonlighting proteins are listed in the moonlighting data base website (http://moonlightingproteins.org/), and the number will be further increased in the future. Moonlighting is common in bacteria; additionally, most of the currently known moonlighting proteins and their functions have been observed in Gram-positive bacteria.5 This might be in relation to their less complex cell wall structure, resulting in easier protein translocation.6 Although moonlighting proteins have been studied relatively well in bacteria, growing evidence indicates that moonlighting proteins are also common in eukaryotic cells and the significance of these multiple-roles proteins should be examined in detail to understand the complexity of human diseases, as previously reported in tumorigenesis.7 Among the eukaryotic moonlighting proteins, secreted moonlighting proteins are actively involved in multiple functions because these proteins translocate intercellularly and affect multiple activities. Sequences of moonlighting proteins do not contain any specific known motifs for translocation, export, import, or anchoring.6 The underlying mechanisms of how the secreted moonlighting proteins are regulated for translocation depend on their activity, compared to secreted non-moonlighting proteins, in the cells have yet to be fully understood.
In this review, we focused on proteins secreted in eukaryotic cells with moonlighting properties: functions other than their conventional role in the cell at different locations in response to inflammation. Chronic inflammation contributes to the development of many diseases, including cancer, arthritis, heart disease, and Alzheimer's disease.89 Cells including stromal cells, immune cells, and stem cells are important mediators of the inflammatory environment. When the contribution of the inflammatory environment becomes evident, secreted proteins from these cells play an important role in the inflammation process. Therefore, many secreted proteins exhibiting multifunctional activities could influence several pathways, indicating a highly adapting moonlighting role in the inflammation process. Recent studies on secreted proteins in intercellular translocation have enhanced our understanding of their roles in cancer and inflammation, although the mechanism of action and the multiple physiological functions of the secreted proteins are yet to be fully understood. In this review, we summarize recent studies characterizing the moonlighting features and multi-biological roles of the secreted proteins, in addition to highlighting the potential implications of these proteins in inflammation.
SECRETED MOONLIGHTING PROTEINS IN ANTI-INFLAMMATORY SIGNATURE
Thymosin beta-4
Thymosin beta-4 (Tβ4) is a highly conserved secreted 43-amino acid peptide that is found in all tissues and in all cell types. The full-length Tβ4 polypeptide has been shown to be effective in reducing inflammation and to be associated with wound healing, hair growth, immunomodulation, and angiogenesis.10 It is also reported that Tβ4 is induced by several nonsteroidal anti-inflammatory drugs,11 supporting its role in anti-inflammation. Moreover, Tβ4 polypeptide down-regulated H2O2-triggered activation of the extracellular Signal-regulated Kinase and Jun N-terminal kinase/mitogen-activated protein kinase, as well as nuclear factor kappa light chain enhancer of activated B cells, suggesting that these signaling pathways might be involved in the anti-inflammatory effects of Tβ4 activation.12 Exogenous and secreted Tβ4 has anti-inflammatory effects in a bleomycin-induced mouse model of lung fibrosis13 and many other inflammatory diseases.1214 Overall, Tβ4 exhibits a general role in anti-inflammation and further suggests a variety of possible therapeutic applications.
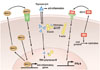
Although conditioned media of cells transfected with Tβ4 has been used to detect Tβ4 using Western blot15 and its major anti-inflammatory activity in the cells of many multicellular animals has been recognized, Tβ4 can bind globular actin (G-actin). Consequently, Tβ4 could be involved in cytoskeletal regulation by inhibiting the polymerization of G-actin into fibrous actin (F-actin)16 (Fig. 1). Because of its small size, Tβ4 might even enter the nucleus by diffusion through nuclear pores. Although the nucleus contains several other actin-binding proteins and significant amounts of G-actin, the exact function of Tβ4 in the nucleus is unknown. Recent data suggest that the interaction of Tβ4 with nucleolar actin might contribute in modulating the transcription activity of the RNA polymerases,17 indicating that it might act as another transcription factor.18 Indeed, it has been reported that Tβ4 upregulates miR146a to suppress the toll-like receptor in the pro-inflammatory pathway.19
Besides moonlighting activity, some of the multiple activities of Tβ4 unrelated to actin might be mediated by a tetrapeptide enzymatically-cleaved from its N-terminus, N-acetyl-ser-asplys-pro (Ac-SDKP), best known as an inhibitor of the proliferation of hematopoietic stem cells in bone marrow and a promoter of angiogenesis in vitro and in vivo in various models.20 Although diverse extracellular effects of Tβ4 have been demonstrated, regulation of its synthesis and secretion to the extracellular environment needs to be elucidated in detail.
Nonsteroidal-anti-inflammatory drug activated gene-1/growth differentiation factor 15
Nonsteroidal-anti-inflammatory drug activated gene-1 (NAG-1) is belonging to the transforming growth factor-beta (TGF-beta) superfamily and is expressed quite widely; however, under normal conditions, the placenta, colon, and prostate are the only tissues that express large quantities of NAG-1 transcripts.21 Furthermore, all the tissues might be influenced by NAG-1 because NAG-1 is known to circulate in the bloodstream.22 The biological role of secreted NAG-1 in many diseases remains poorly understood, sometimes with contradictory evidences. NAG-1 might share at least some common functions with other members of the TGFβ superfamily cytokines. For instance, NAG-1 expression reduces TNF-α secretion in macrophages23 and inhibits the proliferation of the primitive hematopoietic progenitors24 and epithelial cancer cell lines.2526272829 In contrast to other TGF-β superfamily genes, several nonsteroidal antiinflammatory drugs and dietary anti-inflammatory compounds induce NAG-1,21 indicating its role in anti-inflammation. Indeed, overexpressed NAG-1 protein in NAG-1Tg+/FVB mice (Friend leukemia virus B background) inhibited the formation of urethane-induced lung tumors and inflammation through down-regulation of the p38MAPK signaling pathway, and induced apoptosis through the activation of caspase 3/7.30 Interestingly, we also found that compared to control mice, NAG-1Tg+/BL6 mice were less sensitive to dextran sodium sulfate induced colitis,31 suggesting that NAG-1 has anti-inflammatory activity in the intestine as well. These results indicate that NAG-1 not only plays a role in suppressing inflammation but also in suppressing inflammation-induced tumorigenesis in vivo. Furthermore, we reported that NAG-1 modulates metabolic activity by increasing the expression of key thermogenic and lipolytic genes in the adipose tissue.22 The study also suggested that NAG-1 is a novel therapeutic target of potential use in preventing and treating obesity and insulin resistance, wherein inflammation plays a critical role. The serum concentration of NAG-1 from human NAG-1 expressing xenografts ranges from 20–30 ng/mL, whereas NAG-1Tg+/BL6 mice exhibit 50–80 ng/mL.22 Together, these data indicate that NAG-1 is an excellent target for innovative foundational research aimed at understanding, treating, and preventing cancer, inflammation, and/or obesity.
We recently reported that pro-NAG-1 is expressed in the nucleus and controls transcriptional regulation.32 Nuclear NAG-1 affects the SMAD pathway by inhibiting the binding activity of SMAD to target genes. Blocking the nuclear export of NAG-1 by exportin 1 inhibitor results in the inhibition of the secretion of NAG-1 to the extracellular matrix (ECM). These findings warrant the need for further study of the molecular mechanisms by which one protein translocates from the nucleus to the ECM, affecting anti-inflammation activity through protein production, modification, and translocation.
Because the receptor for NAG-1 expressed only in brain has been identified by several groups,33343536 it is necessary to elucidate which NAG-1 (secreted or nuclear) possesses anti-inflammatory activity. However, it is clear that nuclear NAG-1 contain anti-cancer activity as seen in the transgenic mice, whereas secreted NAG-1 may have a pro-cancer property.32
Interferon-stimulated gene 15
Another fascinating moonlighting protein with a key inflammatory homoeostatic function is the ubiquitin analogue interferon-stimulated gene 15 (ISG15), which is one of the most upregulated genes upon type I interferon (IFN-I) treatment or pathogen infection.37 It has been reported that the primary effect of extracellular ISG15 is the stimulation of IFN-γ production37 and may regulate multiple aspects of the host immune response to pathogens.38 This suggest that extracellular ISG15 may regulate infection and inflammatory processes involving IFN-I signaling. Since IFN-I may exert anti-inflammatory effects, a combined ex vivo and in vitro approach recently uncovered a novel extracellular ISG15-mediated anti-inflammatory signaling cascade, including IL-10.39
On the contrary, the primary intercellular function of ISG15 protein is ISGylation, which is able to add covalent bond to the cytoplasmic and nuclear proteins, similar to ubiquitination. The ISGylation process utilizes ubiquitin like modifier activating enzyme 7 as E1 enzyme, ubiquitin conjugating enzyme E2 L6 as E2 enzyme, and EFP/HERC5 as E3 enzyme.4041 Given that it expresses at multiple cellular locations; it is clear that ISG15 has multiple functions. It is suggested that extracellular ISG15 is involved in IFN-γ production,37 whereas intercellular ISG15 plays an important role during viral infection (Fig. 1).42 In addition, intercellular ISG15 is IFN-α/β-inducible to ensure the Ubiquitin Specific Peptidase 18-dependent regulation of IFN-α/β and prevention of IFN-α/β-dependent auto inflammation, instead of acting as a substrate for ISGylationdependent antiviral immunity.43 These results suggest that human intracellular ISG15 is not only redundant for viral infection but also a key negative regulator of IFN-α/β-based immune response.
SECRETED MOONLIGHTING PROTEINS IN PRO-INFLAMMATORY SIGNATURE
Matrix metalloproteinase-12
Matrix metalloproteinase-12 (MMP-12), also called macrophage metalloelastase, was originally detected in the alveolar macrophages of cigarette smokers.44 It is secreted as a 54-kDa inactive pro-enzyme that is activated by the proteolytic cleavage of the pro-domain, followed by processing into two active enzymes of 45 kDa and 22 kDa, respectively. MMP-12 degrades ECM proteins, including elastin, type IV collagen, fibronectin, laminin, and gelatin, and is involved in the turnover of the matrix, cell migration, tissue repairing, and remodeling.45 Pro-inflammatory roles of MMP-12 in inflammation and immunity are known. Clinical and experimental studies have shown that MMP-12 is actively involved in the pathogenesis of inflammatory diseases, such as chronic obstructive pulmonary disease.46 For example, MMP-12 modulates high fat dietinduced glomerular fibrogenesis and inflammation in a mouse model with obesity47 and overexpression of MMP-12 is associated with increased macrophage migration in cutaneous granulomas and enhanced inflammatory arthritis in transgenic rabbits.4849 In addition, inhibition of MMP-12 with a small molecule has been found to reduce the inflammatory process associated with exposure to smoking cigarette condensates, strongly suggesting a specific involvement of MMP-12 in lung inflammation. Hence, MMP-12 exhibits pro-inflammatory property.
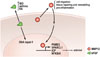
Interestingly, the secreted MMP-12 is taken up by cells as a cis isomer and is localized in the nucleus. Nuclear MMP-12 increases the NFKB inhibitor alpha transcription, which is essential for optimal IFN-α secretion and antiviral immunity.50 The antiviral response is tightly regulated by MMP-12 through DNA binding of multiple gene exons, including those encoding proteasome activator complex subunit 3, the immunoproteasome cap protein, and SPARCL1, which reduces their mRNA and protein levels in vitro and in vivo in Mmp12-/- mice. Although MMP-12 likely has a role in inflammatory activity in the ECM, its nuclear activity contributes to the anti-viral response (Fig. 2).
Basic fibroblast growth factor
Basic fibroblast growth factor (bFGF or FGF-2) is a potent angiogenic factor and an endothelial cell mitogen. The bFGF protein synergistically also potentiates the inflammatory mediator-induced leukocyte recruitment, at least in part, by enhancing the up-regulation of the cell adhesion molecules in the endothelium.51 The expression and secretion of the angiogenic factors, including bFGF, is increased at chronic inflammation sites, and the bFGF protein is increased in the serum and affected tissue of the patients with rheumatoid arthritis, inflammatory bowel disease, or asthma.52 In addition, recent data suggest that bFGF cooperates with the pro-inflammatory cytokine IL-17 to induce inflammatory response.53 Therefore, secreted bFGF plays a role during both inflammation and angiogenesis.
In contrast to the pro-angiogenic and pro-inflammatory activity of bFGF in the ECM, the nuclear forms of bFGF trigger a mitogenic stimulus under serum starvation conditions, and do not specifically protect the cells from apoptosis.54 Nuclear bFGF-mediated chemo-resistance is also associated with increased DNA-dependent protein kinase expression and accelerated DNA repair.55 In addition, nuclear bFGF has contributed to drug resistance following standard chemotherapy treatment in a subset of triple negative breast cancers.56 Besides its angiogenesis and inflammatory activity, nuclear bFGF contributes to pro-tumorigenic activity.
CONCLUSION
Protein moonlighting is gradually being established and more proteins with moonlighting functions will certainly be identified. In upcoming reviews, many more secreted moonlighting proteins will likely be identified. We can initiate developing hypotheses on the role of moonlighting proteins in inflammation only after an increased number of such proteins has been identified. Furthermore, the knowledge of general medicine will be enhanced upon the elucidation of the role of moonlighting proteins. The current therapy of inflammatory diseases is based on the suppression of pro-inflammatory proteins or the induction of anti-inflammatory proteins. Studying moonlighting activity including protein translocation in the cell may provide new concepts with which to treat inflammatory diseases, as the protein could stay longer in one location where the protein has an anti-inflammatory activity (Table 1). For instance, Tβ4 has anti-inflammation activity in the ECM; therefore, facilitating Tβ4 movement from the cytoplasm to the ECM may favor anti-inflammatory activity. Explicit questions that will be addressed to explain the complexity of humans and unexplained diseases are as follows: 1) What is the molecular mechanism underlying the evolution of moonlighting proteins? 2) What is the common domain that can be found in both secreted and non-secreted moonlighting proteins? 3) Does protein moonlighting add complexity to the existing systems in the cell? 4) What are the molecular mechanisms that moonlighting proteins use to translocate intercellularly and intracellularly? 5) What is the significance of moonlighting proteins in human disease? 6) How many moonlighting proteins are likely to be therapeutic agents? 7) Does protein moonlighting contribute to adverse drug responses?
Hopefully, answers to these questions can be reached within the next decade. Assuming that most secreted proteins possess moonlighting activity, this assumption will lead us to further understanding molecular mechanisms of human diseases.

 XML Download
XML Download