

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Intellectual disability (ID) is characterized by impairment of cognitive and adaptive functions, with onset before age 18 years. ID is usually identified during infancy or early childhood because of developmental delay (DD). However, ID is formally diagnosed upon obtaining an intelligence quotient (IQ) score of less than 70.12 ID occurs in approximately 1– 3% of the population, and is largely caused by genetic abnormalities.1 However, it has extensive genetic heterogeneity, and 60% of the cases of ID still do not have a known etiology.3 ID is often accompanied by clinical features of dysmorphism, congenital anomalies, or epilepsy. Chromosomal microarray (CMA), with a higher diagnostic yield (15– 20%) than that of the G-banded karyotype (~3%), is now a first-titer clinical diagnostic test for individuals with unexplained DD/ID, autism, or multiple congenital anomalies.4 CMA has a 100-fold higher resolution than G-banded karyotype, and detects submicroscopic copy number variants (CNVs) across the entire genome. In many cases, the detected CNVs are disease-causing genomic alterations, although benign variants or variants of uncertain significance (VOUS) are also frequent. Although CMA and clinical data have extended the spectrum of understanding the genetic causes of ID, interpretation thereof and appropriate management, including genetic counseling, remain problems in a clinical setting.
Here, we present our CMA results with clinical data in patients with unexplained DD/ID accompanying dysmorphism, congenital anomalies, failure to thrive (FTT), or epilepsy. We aimed to describe characteristics of clinical features in patients with pathogenic CNVs.
Go to : 
MATERIALS AND METHODS
Patients
We collected clinical and CMA data of the patients who visited Konyang University Hospital for evaluation of unexplained DD/ID in a period of one year. In total, 50 patients who had taken the CMA test to evaluate the etiology of unexplained DD/ID between September 2013 and October 2014 were included. DD/ID was defined by IQ lower than 70 or developmental quotient lower than 85. All the patients had severe DD/ID with or without congenital anomalies, growth failure, or epilepsy. Exclusion criteria included 1) brain damage owing to hypoxic ischemic encephalopathy, periventricular leukomalacia, intracranial hemorrhage, infarction, or sequeale of encephalitis; 2) metabolic abnormalities, such as hypothyroidism, organic acidemia, amino acidopathy, peroxisomal disorder, etc.; and 3) recognizable chromosomal syndromes or single gene disorders, such as Down syndrome, Klinefelter syndrome, or Fragile-X syndrome. Phenotypes were described as follows. 1) Dysmorphism: anatomical structures or their measures are outside the normal range. 2) Major organ anomalies: CNS, heart, and uro-genital anomalies were included. 3) FTT: height and weight growth lie below the third percentile. 4) Microcephaly: head circumference is below third percentile. 5) Macrocephaly: head circumference is above the 97th percentile. 6) Epilepsy: recurrent seizure disorder with abnormal EEG. 7) Autism: neurodevelopmental disorder characterized by impaired social interaction, impaired verbal and non-verbal communication, and restricted and repetitive behavior.
Chromosomal microarray
DNA was extracted from peripheral blood leukocytes. CMA analysis was performed using CytoScan 750K (Affymetrix, Santa Clara, CA, USA). The array is characterized with >750436 CNV markers, including 200436 genotype-able SNP probes and >550000 non-polymorphism probes. The overall average marker space is 4127 base pairs. All data were visualized and analyzed with the Chromosome Analysis Suite (ChAS) software package (Affymetrix) using Human Genome build hg19. This software for CytoScan 750K was designed to detect a minimal size of 200 kb aberrations.
Interpretation
All detected CNVs were compared with known CNVs databases, such as the Database of Genomic Variants (http://dgv.tcag.ca), University of California Santa Cruz Genome Browser (http://genome.ucsc.edu), and DECIPHER (http://decipher.sanger.ac.uk). In cases of potentially significant CNVs not listed in the above databases, literature searches in the PubMed database was performed. We classified CNVs as pathogenic, benign, or VOUS based on literature guidelines.3
Statistical analysis
Statistical analyses were performed using SPSS 19.0 (IBM Corp., Armonk, NY, USA). Clinical features of dysmorphism, FTT, microcephaly, and epilepsy in patients with pathogenic CNVs and normal CMA were compared using the chi-squared test. Fischer's exact test was used in cases where expected numbers of the patients were below 5, such as in the clinical features of CNS anomaly, heart anomaly, uro-genital anomaly, macrocephaly, and autism. A p-value ≤0.05 was considered to indicate statistical significance.
Ethics statement
Approval was obtained from the Konyang University Hospital Institutional Review Board (2015-07-012-002). The title of the approved study is “Clinical utility of chromosomal microarray in patients with unexplained developmental delay/intellectual disability.” Written informed consent was obtained from the parents/legal guardians of all the participants. Written informed consent was about confirming that participants or parents/legal guardians understand CMA tests and agree for the genetic test and for their participation in human materials research. Konyang University Hospital Institutional Review Board approved this consent procedure.
Go to :
RESULTS
CMA was performed in 50 patients, comprising 26 males and 24 females. The mean age at the time of study was 5.4±5.9 years (range: 0.1– 32 years). All the patients had DD/ID. The other most common clinical features were dysmorphism in 39 patients, FTT in 27 patients, epilepsy in 19 patients, major organ anomalies in 12 patients, and autism in 9 patients (Table 1). We identified 37 CNVs in 29 of the 50 (58.0%) patients. Among the 29 patients with abnormal CMA results, the CMA analysis of the parents was done for 16 patients, and a siblings study was done for one patient. The inheritance pattern consisted of eight de novo sporadic cases and nine familial cases. Paternal origin was observed in six patients and maternal origin in three patients. The CMA database and parental findings categorized the abnormal CMA results as pathogenic CNVs in 18/29 (62.1%) patients, benign CNVs in 6/29 (20.7%), and VOUS in 5/29 (17.2%) patients. The diagnostic yield of detecting pathogenic CNVs was 18/50 (36.0%). The CMA results and clinical features are summarized in Table 2. Five out of 37 CNVs were sex chromosome rearrangements, 21 were duplications, and 16 were deletions. The size of the CNVs ranged from 227 kb to 18 Mb.
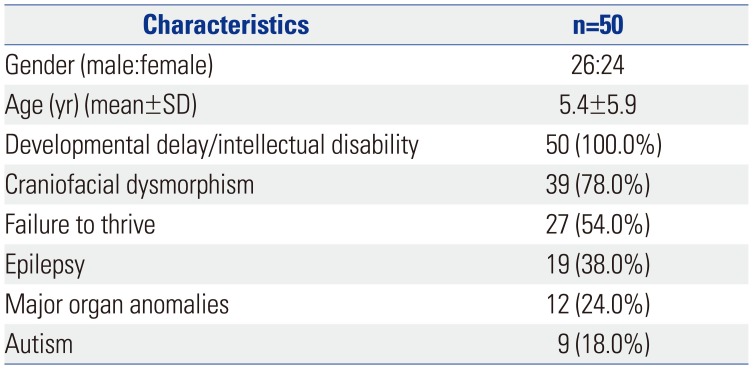
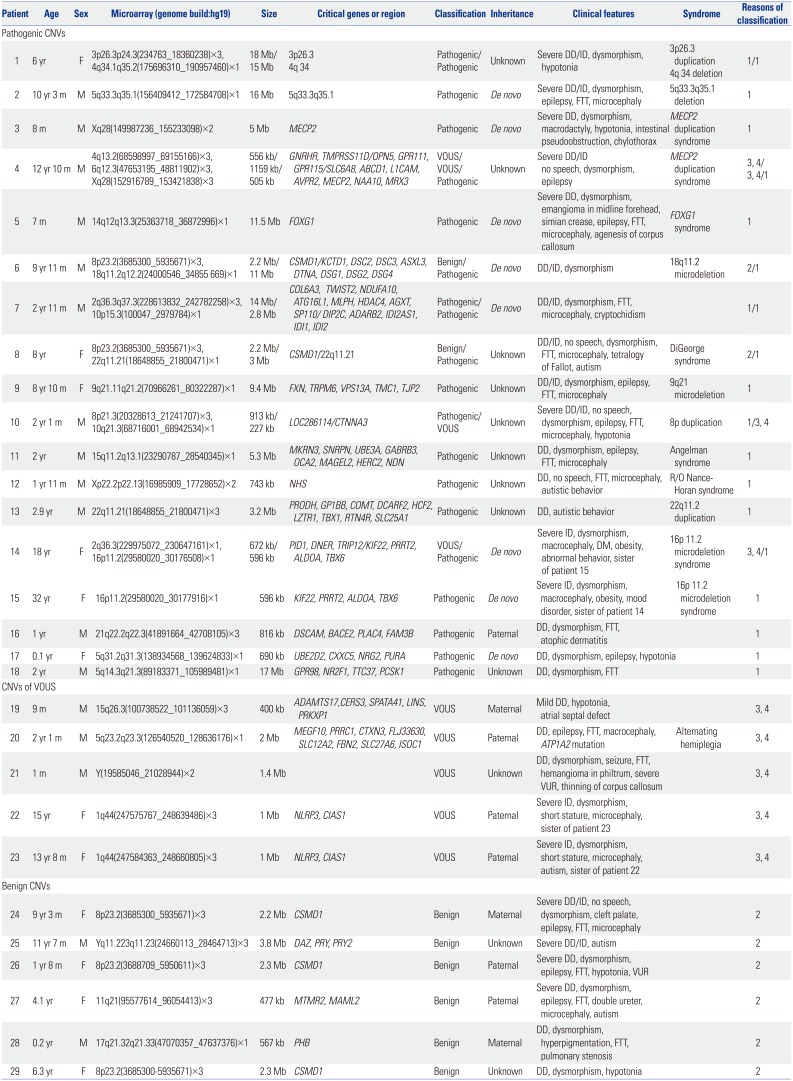
Table 1
Clinical Characteristics of the 50 Patients
![]()
Table 2
Clinical and Genetic Features in 29 Patients with CNVs
CNVs, copy number variants; VOUS, variants of uncertain significance; CMA, chromosomal microarray; DD, developmental delay; ID, intellectual disability; FTT, failure to thrive; DM, diabetes millitus; VUR, vesicoureteral reflux.
1. Overlapping with a known imbalance syndrome; 2. In the category of genomic imbalance in healthy individuals as per public database; 3. CNV is not a common polymorphism; 4. Genes in the CNV are not associated with patient's phenotype.
![]()
Clinically known syndromes, such as the MECP2 duplication syndrome, FOXG1 syndrome, DiGeorge syndrome, Angelman syndrome, Nance-Horan syndrome, and other known microdeletion syndromes, were identified. DiGeorge syndrome could be diagnosed without the CMA test because of its typical phenotype. However, the patient was suspected to have additional genomic alterations because of severe language impairment and autistic behavior. The results showed a 22q11.2 deletion and benign CNVs of 8p23.2 duplication. Angelman syndrome in patient 11 was finally confirmed by methylation PCR after CMA test. In this study, 14 patients presented reported rare CNVs (Table 2).
Among the nine familial patients, eight patients were found in VOUS and one patient (patient 16) was in pathogenic CNVs. In the eight patients in VOUS, all the parents were normal, whereas the father of patient 16 manifested a mild phenotype, such as mild atophic dermatitis and low body weight during childhood.
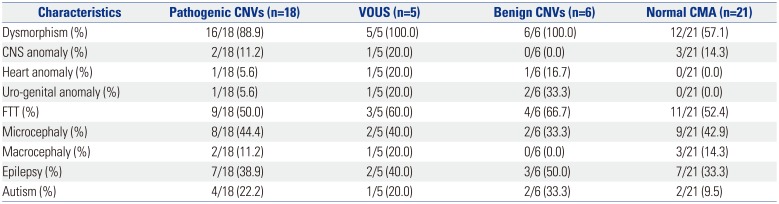
Further, phenotypes among the patients with pathogenic CNVs, VOUS, benign CNVs, and normal CMA were compared (Table 3). The clinical features most often seen in patients with pathogenic CNVs were dysmorphism (88.9%), FTT (50.0%), microcephaly (44.4%), epilepsy (38.9%), major organ anomaly (22.4%), and autism (22.2%), in the order of frequency. Patients with normal CNVs presented dysmorphism (57.1%), FTT (52.4%), microcephaly (42.9%), epilepsy (33.3%), major organ anomaly (14.3%), and autism (9.5%). Dysmorphism (p=0.028) was significantly more frequent in patients with pathogenic CNVs than in those with normal CMA (Fig. 1). Autism (p=0.387), epilepsy (p=0.718), and microcephaly (p=0.921) were more frequent in patients with pathogenic CNVs than in patients with normal CMA, although the difference was not significant (Fig. 1).
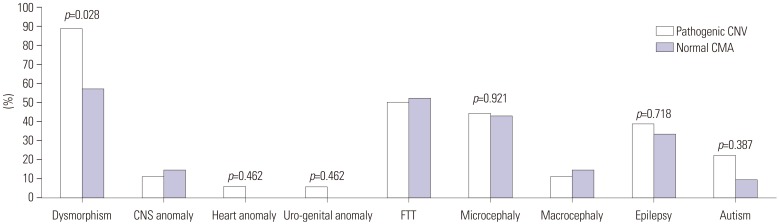 | Fig. 1Comparison of phenotypes between the patients with pathogenic CNVs and normal CMA. Dysmorphism (p=0.028) was significantly more frequent in patients with pathogenic CNVs than in those with normal CMA. Autism (p=0.387), epilepsy (p=0.718), and microcephaly (p=0.921) were more frequent in patients with pathogenic CNVs than in patients with normal CMA, but the difference was not significant. CNVs, copy number variants; VOUS, variants of uncertain significance; CMA, chromosomal microarray; CNS, central nervous system; FTT, failure to thrive.
|
Table 3
Percentage of Presented Clinical Manifestations in Different CNVs Groups
![]()
Among the nine clinical features listed in Table 3, patients with normal CMA presented 2.2±1.3 (range: 1– 4) manifestations, and patients with pathogenic CMA presented 2.8±1.3 (range: 1– 5) manifestations. Two or more symptoms were presented by 61.9% (13/21) of the patients with normal CMA and by 83.3% (15/18) of the patients with pathogenic CMA.
Go to :
DISCUSSION
In this study, we identified chromosomal imbalances in 58.0% (29/50) of our patients. Of these, pathogenic CNVs were found in 62.1% (18/29), benign CNVs in 20.7% (6/29), and VOUS in 17.2% (5/29) of the patients. The overall diagnostic yield was, therefore, 36.0% (18/50). In recent reports, the diagnostic yield of CMA was shown to have increased up to 10– 28% for genetic testing in patients with unexplained DD/ID, autism spectrum disorders, or multiple congenital anomalies, as compared to 3% for G-banded karyotype.4567891011 In our study, the diagnostic yield was higher than that found in previous reports with similar patients. In Korea, the cost of CMA is not covered by Korean Health Insurance; therefore, patients for the CMA test were very carefully selected by a clinical geneticist, Hyon J. Kim. The correct diagnosis included the MECP2 duplication syndrome, FOXG1 syndrome, Angelman syndrome, and other known microdeletion or microduplication syndromes. In these cases, the diagnosis could be useful for predicting the clinical progress, preparing for symptoms not yet presented, and deciding the plan for the evaluation and management of the disease. Furthermore, using a familial study, we can estimate a recurrent risk in the family and can give more information about the disease. A previous large-scale study showed that 54% of the abnormal variants generated a recommendation for clinical action.12 The CMA test is an important diagnostic test that influences medical management. It also influences medical management in cases of VOUS, although clinicians should periodically review updated information in order to provide appropriate medical management. As data from genetic studies increase, CNV interpretation and useful information can be updated. Palmer, et al.13 reported that interpretation of CMA could change over time. For example, patients 22 and 23 sisters showed CNV of VOUS until now; however, the interpretation of the 1q44 trisomy in these patients can change over time.
In our study, among the 21 patients who showed no abnormalities in the CMA test, three patients were further tested using whole exome sequencing. Two familial patients with ID, severe short stature, and macrodontia were diagnosed with the KBG syndrome, with confirmed ANKRD11 mutation.14 The other patient was diagnosed with Pitt-Hopkins syndrome, with confirmed missense mutation in TCF4. In cases where the CMA test did not allow diagnosis, whole exome or whole genome sequencing could identify the genetic causes of ID. Finally, genome sequencing could be applied as a single genetic test, with a higher diagnostic yield, in the majority of patients with severe ID.15
Some researchers have investigated the characteristics of CNVs in patients with DD/ID. Patients with ID and multiple congenital anomalies show higher burden of CNVs than those with ID alone.16 DD/ID risk is known to increase in relation to CNV size and craniofacial anomalies. Further, cardiovascular defects are enriched for large CNVs relative to epilepsy and autism spectrum disorder.17 Multiple large CNVs, including CNVs of unknown significance, are known to result in severe clinical presentation.18
We investigated the relation between other phenotypes and pathogenic CNV in patients with DD/ID. Dysmorphism (p=0.028) alone was significantly more frequent in patients with pathogenic CNVs than in those patients with normal CMA. A previous study showed that congenital anomalies, microcephaly, short stature, and FTT were more frequent in children with pathogenic CNVs.19 However, in our study, such phenotypes were not different between the two groups, because most of the patients with normal CNVs were also in the spectrum of genetic variation that have yet to be identified by further testing, such as whole exome sequencing. Therefore, most of the phenotypes, such as congenital anomalies, microcephaly, and FTT were as frequent in patients with normal CMA as in patients with pathogenic CNVs. The number of phenotypes, however, was different. A greater number of patients with pathogenic CMA, compared to those with normal CMA, presented two or more symptoms. We can, therefore, expect that in patients with unexplained DD/ID, the pathogenic CNV might be more frequently found if the patients have two or more phenotypes in addition to DD/ID.
Our results emphasize the importance of the CMA test in the clinical evaluation of patients with unexplained DD/ID. This study was limited to a small number of patients; thus, diverse statistical analysis was not possible. However, our data involving detailed phenotype analysis and CNVs can add to databases for future genetic studies for discovering new candidate genes and molecular pathways underlying unexplained DD/ID.
Go to :
 XML Download
XML Download