

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Multiple myeloma (MM) is a devastating plasma cell malignancy characterized by the accumulation of clonal proliferation of neoplastic plasma cells throughout the bone marrow.1 MM represents 1% of all cancers and is responsible for 10% of all hematological neoplastic diseases, making it the second most common blood cancer.2 Growing evidence suggests that the microenvironment of bone marrow and cytokines, such as interleukin (IL)-6 and tumor necrosis factor (TNF)-α, plays pivotal roles in the growth and survival of MM cells and influences the clinical manifestation and prognosis of this disease.3 Despite improvements in current therapies for MM, including chemotherapy and stem cell transplantation, MM is still regarded as an incurable disease, with a 5-year survival rate of approximately 45%.4 Therefore, there is a pressing medical requirement for identifying novel alternative approaches and illustrating molecular mechanisms to improve the prognosis of and therapeutic effects in patients with MM.
The presence of monoclonal immunoglobulins and defective innate or adaptive immune responses makes MM patients vulnerable to bacterial, fungal, and viral infections, which remain a major cause of death, along with general organ failure in patients suffering from MM.5 During an infection, activation of an immune response depends on the identification of pathogen components by a family of pattern recognition receptors (PRRs), including toll-like receptors (TLRs).6 Upon recognition, TLRs bind to adaptors to activate intracellular signal transduction pathways that culminate in the activation of two critical transcriptional factors: interferon regulatory factor 3 (IRF3) and NF-κB, which are pivotal for the induction of interferons (IFNs) and proinflammatory cytokines/chemokines.78 TLR3 is a receptor that is exclusively mediated by the Toll-IL-1 receptor (TIR) domain-containing adaptor inducing IFN-β (TRIF) adaptor, participating in the recognition of double-stranded RNA (dsRNA) from viruses and of mammalian RNA from necrotic cells.9 TLRs have been reported to be highly and heterogeneously expressed on freshly isolated myeloma cells and MM cell lines and involved in tumor growth and host immune response.1011
Tripartite-motif (TRIM) protein family, consisting of more than 70 members, are implicated in diverse functions in a wide range of biological processes, including cell growth, differentiation, development, apoptosis, and immunity.1213 Structurally, TRIM family is characterized by highly conserved N-terminal tripartite (also known as RBCC) motifs (a RING finger, one or two B boxes, coiled-coil domains) and a variable C-terminal half that determines the function specificity of several TRIM proteins.13 It has been proposed that TRIM proteins exhibit RING-finger E3 ubiquitin ligase activity that mediates the posttranslational modification of different substrates.14 Recently, TRIM proteins have been disclosed to positively or negatively regulate carcinogenesis.15 Tripartite-motif-containing protein 56 (TRIM56, also referred to as T56), a novel actor in innate antiviral immunity, is suggested to be an IFN- and virus-inducible factor possessing antiviral activity, mainly depending on its E3 ubiquitin ligase activity and C-terminal structural integrity.16 TRIM56 promotes TLR3-mediated antiviral signaling pathways toward IFN induction through E3 ligase-independent mechanisms.17 In addition, it was reported that TLR3 ligand could strengthen immunity against MM cells or directly trigger cell apoptosis, while TLR antagonists are able to promote MM survival, proliferation, and immune escape.6 However, whether TRIM56 could be a regulator of TLR3 in MM cells remains unknown.
In the present study, we first investigated the expression and biological function of TRIM56 in MM cells. Furthermore, we explored whether TRIM56 participated in the development of MM by regulating TLR3 signaling pathway.
MATERIALS AND METHODS
Cell lines and culture medium
Human MM cell lines (NCI-H929, RPMI-8266, LP-1, OPM2 and U266) were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). All cell lines were cultured in RPMI 1640 (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA), 2 mM L-glutamine, and 1% streptomycin/penicillin, at 37℃ with 5% CO2. Normal plasma cells (PCs, CD138+) were isolated and purified from normal healthy donors as described previously.18 This study was approved by the Ethical Committees of The First Affiliated Hospital of Xi'an Jiao Tong University.
Cell transfection
Small interfering RNAs (siRNAs) against TRIM56 (T56si-1 and T56si-2) and negative control (si-control) were synthesized from GenePharma (Shanghai, China). Poly (I:C) (a dsRNA surrogate) and poly (dA:dT) (a dsDNA surrogate) were purchased from Sigma (Sigma, St. Louis, MO, USA). Transfection with sicontrol, T56si-1, T56si-2, poly (dA:dT), or poly (I:C) into RPMI8226 or U266 cells was performed using Lipofectamine 2000 Reagent (Invitrogen) according to the manufacturer's instructions.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from MM cell samples using TRIzol reagent (Invitrogen). The first strand cDNA was synthesized from total RNA (1 µg) using PrimeScript RT reagent kit with gDNA Eraser (Takara Bio, Dalian, China). The expression level of TRIM56 mRNA was detected by an ABI 7500 real-time PCR instrument (Applied Biosystems, Foster City, CA, USA) using the SYBR Green PCR kit (Takara Bio) and normalized to GAPDH. The relative expression of TRIM56 was calculated by the 2−ΔΔCT method. The sequences of sense and antisense primers used were: GAPDH: 5′-GACACCCACTCCTCCACCT-3′ and 5′-ATGAGGTCCACCACCCTGT-3′; TRIM56: 5′-CTGCTTG GMGACTTCCTGAC-3′ and 5′-GTGGATGGTSCCGTTACTG AG-3′
Western blot analysis
Total protein lysates were extracted from MM cells by RIPA lysis buffer (Beyotime, Jiangsu, China) with EDTA-free Protease Inhibitor Cocktail (Roche, Mannheim, Germany) and quantified using an Enhanced BCA protein assay kit (Beyotime). Equal amounts of total protein (10 µg) were fractionated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (Millipore, Bedford, MA, USA) overnight. After being blocked with 5% non-fat dry milk in tris-buffered saline with Tween 20, the membranes were blotted overnight at 4℃ with the following primary antibodies: TRIM56 (1:1000; Cell Signaling Technology, Danvers, MA, USA), TLR3 (1:500; Cell Signaling Technology), TRIF (1:1500; Cell Signaling Technology), RIP1 (1:500; Cell Signaling Technology), followed by incubation with horseradish peroxidase-conjugated secondary antibody anti-mouse IgG (1:2000; Millipore) for 1 h. Protein signals were visualized by enhanced chemiluminescence (Thermo Scientific, Waltham, MA, USA).
Cell viability assay
Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay according to the manufacturer's protocol. In brief, MM cells were seeded in 96-well plates at a density of 3×103 /well. At indicated time points, 20 µL of MTT (0.5 mg/mL, Sigma) was added to each well for further incubation for 4 h at 37℃. Following removal of the medium, 150 µL of DMSO (Sigma) was added to dissolve the crystals, and absorbance was measured at 450 nm on a microplate reader (Bio-Tec, Winooski, VT, USA).
Apoptosis analysis by flow cytometry
Apoptosis of MM cells were detected using an Annexin V-FITC Apoptosis Detection Kit (BD Pharmingen, Heidelberg, Germany), following the instructions described by the manufacturer. The samples were analyzed with a Fluorescence Activated Cell Sorter (FACS) Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA). 2 µM arsenic trioxide (ATO, As2O3) was applied to induce apoptosis of MM cells.19
Caspase3 activity assay
The relative caspase3 activity of MM cells was detected using the Caspase3 Colorimetric Assay Kit (Beyotime) according to the manufacturer's instructions, and samples were detected at the wavelength of 405 nm using a microplate reader (Bio-Tec).
Enzyme-linked immunosorbent assay
The concentrations of IFN-β, IL-6, and TNF-α in the supernatant of RPMI8226 or U266 cells were measured using respective ELISA kits (Xitang Company, Shanghai, China).
Statistical analysis
All numerical data are expressed as means±standard deviations. Comparisons between two or more groups were analyzed by one-way analysis of variance or Student's t-test using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Statistical significance was determined with data from at least three independent experiments, and p<0.05 was considered statistically significant.
RESULTS
TRIM56 expression down-regulated in MM cells
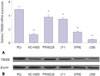
The expression level of TRIM56 mRNA and protein in normal PCs and MM cell lines was firstly determined by qRT-PCR and western blot. As presented in Fig. 1, TRIM 56 mRNA and protein expressions markedly decreased in MM cells (NCI-H929, RPMI8226, LP-1, OPM2, and U266), compared to normal PCs. Considering the highest expression of TRIM 56 in RPMI8226 and lowest expression in U266 cells, we decided to choose these two MM cells for further analysis. These data indicated that dysregulation of TRIM 56 may be associated with MM pathogenesis.
TRIM56 impedes proliferation and enhances apoptosis in MM cells
To explore the biological function of TRIM56 in the development of MM, we up-regulated TRIM56 expression by stimulating U266 cells with poly (dA:dT), a dsDNA surrogate mediating type I IFN induction. Western blot analysis showed that the protein level of TRIM56 in U266 cells with poly (dA:dT) stimulation was considerably increased, compared with control cells (Fig. 2A). MTT assay revealed that restoration of TRIM56 expression by stimulation with poly (dA:dT) led to an apparent suppression on U266 cell growth (Fig. 2B). Additionally, flow cytometry analysis was performed to assess the effect of TRIM56 on MM cell apoptosis. The results showed that TRIM56 overexpression triggered by poly (dA:dT) stimulation significantly enhanced apoptotic rate of U266 cells (Fig. 2C). Meanwhile, results of caspase3 activity analysis indicated that caspase3 activity was dramatically increased in poly (dA:dT)-treated U266 cells (Fig. 2D). On the contrary, RPMI8226 cells were transfected with siRNAs (T56si-1 or T56si-2) to suppress TRIM56 expression. As expected, transfection of T56si-1 and T56si-2 markedly decreased the protein level of TRIM56 in RPMI8226 cells (Fig. 2E). TRIM56 knockdown by T56si-2 obviously accelerated RPMI8226 cell proliferation (Fig. 2F). Flow cytometry analysis revealed that ATO-induced apoptosis was strikingly suppressed following TRIM56 down-regulation in RPMI8226 cells (Fig. 2G). Consistently, caspase3 activity assay proved that suppressing TRIM56 expression in RPMI8226 cells remarkably attenuated ATO-induced elevation in caspase3 activity (Fig. 2H). These results demonstrated that TRIM56 overexpression hindered cell proliferation and induced apoptosis in MM cells.
TRIM56 induces cytokines secretion in MM cells
We further investigated the effect of TRIM56 on cytokine generation by ELISA in U266 cells treated with poly (dA:dT) or in RPMI8226 cells introduced with T56si-2. As compared with respective control groups, an obvious increase of IFN-α (Fig. 3A), IL-6 (Fig. 3B), and TNF-α (Fig. 3C) concentrations in poly (dA:dT)-transfected U266 cells and a significant reduction of IFN-β (Fig. 3D), IL-6 (Fig. 3E), and TNF-α (Fig. 3F) secretions in T56si-2-transfected RPMI8229 cells were observed, suggesting that TRIM56 overexpression induced the production of inflammatory cytokines.
TRIM56 knockdown suppresses TLR3/TRIF signaling pathway in MM cells
A previous study found that TRIM56 was an essential component of TLR3 antiviral signaling pathway in HeLa cells.17 Therefore, we hypothesized that TRIM56 could also regulate the TLR3 signaling pathway in MM cells. The protein levels of TLR3, TRIF (the cognate adaptor protein of TLR3), and receptor-interacting protein 1 (RIP1, the downstream target of TRIF) were examined by western blot in RPMI8226 cells transfected with T56si-2 or si-control. As expected, TRIM56 knockdown significantly decreased the protein levels of TLR3, TRIF, and RIP1 in RPMI8226 cells (Fig. 4A and B), indicating that TRIM56 positively regulates the TLR3/TRIF signaling pathway in MM cells.
TRIM56 knockdown promotes MM progression by inactivating TLR3/TRIF signaling
To delineate the molecular mechanism of TRIM56 involved in the development of MM, RPMI8226 cells were transfected with T56si-2 or along with poly (I:C), a dsRNA surrogate that could activate the TLR3 signaling pathway.17 MTT assay results revealed that extracellular administration of poly (I:C) effectively abated TRIM56-knockdown-induced increase on RPMI8226 cell viability (Fig. 5A). In addition, ATO-induced apoptosis was significantly decreased in TRIM56-knockdown RPMI8226 cells, whereas cotransfection of T56si-2 and poly (I:C) notably reversed this effect (Fig. 5B). Consistently, in response to TRIM56 knockdown, ATO-induced increase of caspase3 activity was exceptionally repressed in RPMI8226 cells, whereas poly (I:C) treatment significantly abolished the effect of TRIM56 knockdown on caspase3 activity (Fig. 5C). Furthermore, poly (I:C) stimulation dramatically abrogated TRIM56-knockdown-induced decrease in the production of cytokines, including IFN-β (Fig. 5D), IL-6 (Fig. 5E), and TNF-α (Fig. 5F), in RPMI8226 cells. Moreover, western blot analysis demonstrated that decreases in TLR3, TRIF, and RIP1 expression triggered by TRIM56 knockdown was greatly attenuated following treatment with poly (I:C) (Fig. 5G). Together, these results demonstrated that silencing of TRIM56 promotes the progression of MM by blocking TLR3/TRIF signaling pathway.
DISCUSSION
In the present study, we revealed the biological role of TRIM56 and its underlying molecular mechanism in MM. We found that extracellular dsDNA-induced TRIM56 overexpression suppressed the development of MM by impeding MM cell proliferation, elevating inflammatory cytokines secretion, and inducing apoptosis, while TRIM56 knockdown exerted the opposite effects. Importantly, TRIM56 was found to function as a positive regulator of the TLR3/TRIF signaling pathway and activation of TLR3/TRIF signaling pathway abolished the effect of TRIM56 knockdown on the progression of MM.
TRIMs represent a large family of proteins that have been identified to have emerging roles in innate immune system responses to viral infection.13 Notably, some members of TRIM family are expressed in response to type I and II IFNs and participate in antiviral innate immune response through regulating ubiquitination by functioning as E3 ubiquitin ligases.20 Recent studies have reported that several TRIM proteins act as important regulators in many kinds of diseases, including cancer,21 such as TRIM25 in lung cancer,22 TRIM44 in testicular germ cell tumor,23 and TRIM66 in osteosarcoma.24 TRIM56 is an IFN-inducible gene and regulates the antiviral innate immunity. TRIM56 is able to control poly (dA:dT)-mediated pathway, suggesting that TRIM56 may serve as an intracellular sensor for dsDNA.17 The biological role of TRIM56 in MM remains unknown. In the present study, we first found that TRIM56 expression was down-regulated in MM cell lines, compared with normal PCs. Furthermore, poly (dA:dT) stimulation induced high expression of TRIM56 in MM cells, suppressing MM cell growth and inducing apoptosis and inflammatory cytokines production, whereas TRIM56 depletion displayed opposite effects, suggesting that TRIM56 inhibited the tumorigenesis of MM. Consistently, the suppressive role of TRIM proteins has been elucidated in some cancers, such as TRIM26 in lung cancer,25 TRIM16 in melanoma,26 TRIM15 in colon cancer27 and TRIM29 in breast cancer.28
TLRs have been recently recognized as one of PRRs to induce the type I IFN pathway and trigger the initiation of various defense mechanisms by recognizing conserved patterns of microbial structures. Stimulation of TLR signaling induces a range of innate and adaptive immune responses through release of inflammatory cytokines, maturation of dendritic cells, and activation of adaptive immunity.29 TLR3, a major effector of the immune response against viral pathogens, has been found to be frequently expressed in multiple tumors and implicated in cancer progression. For example, TLR3 has been demonstrated to directly trigger apoptosis in human breast cancer cells.30 In hepatocellular carcinoma cells, preferential induction of the apoptotic pathway over the cytokine induction pathway by TLR3 signaling was observed.31 Activation of TLR3 by its ligand poly (I:C) exerted a growth-inhibitory effect against renal cell carcinoma in a TLR3-dependent manner.32 TLR3 agonist poly (I:C), when added after pretreatment with either cycloheximide or type I IFN, is able to induce significant in vitro cell death and proliferation blockade through TLR3 signaling in melanoma tumor cells.33 It has been proposed that TLRs trigger heterogeneous effects on MM primary cells, including increases in proliferation, survival, inflammation, apoptosis induction or prevention, and immune surveillance escape.1134 A previous study demonstrated that TLR3 ligand enhances immunity against MM cells or directly induce cell apoptosis, whereas various TLR antagonists could induce MM survival, proliferation, and immune escape.6 More importantly, TRIM56 was identified as a positive regulator of TLR3 antiviral signaling.17 In line with these reports, our study revealed that TRIM56 knockdown significantly suppressed the TLR3/TRIF signaling pathway. Poly (I:C), a TLR3 agonist, has been demonstrated to induce the expression of inflammatory cytokines and type I IFN, enhancing potent antitumor activity.29 Moreover, transfection with poly (I:C) in RPMI8226 cells dramatically abrogated the positive effects of TRIM56 knockdown on MM cells. These results suggested that TRIM56 suppressed MM carcinogenesis via activating TLR3/TRIF signaling pathway.
In conclusion, TRIM56 overexpression suppressed cell proliferation, induced inflammatory cytokines production, and enhanced apoptosis in MM. Furthermore, the suppressive effect of TRIM56 in MM was mediated by activating TLR3/TRIF signaling pathway. Our findings contribute to a better understanding of the molecular mechanism of TRIM56 involvement in MM development and provide a potential therapy target for preventing MM.




 XML Download
XML Download