

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Olmsted syndrome (OS, OMIM 614594) is a rare genodermatosis characterized by mutilating palmoplantar keratoderma, periorificial keratotic plaques, and severe itching.12 Patients suffer also from diffuse alopecia, constricting digital bands, and onychodystrophy.23 To date, approximately 70 cases of OS have been reported. Most cases are sporadic; however, familial OS has also been found.145 Pathogenic mutations in the transient receptor potential vanilloid 3 gene (TRPV3) are the cause of autosomal-dominant OS. TRPV3 is a thermosensitive cation nonselective channel predominantly expressed in keratinocytes and sensory neurons.6 Gain-of-function mutations in TRPV3 lead to constitutive activity in mutant channels, resulting in enhanced keratinocyte apoptosis and hyperkeratosis.4 Herein, we report a first Korean case of Olmsted syndrome harboring a relatively rare missense mutation within TRPV3, p.Gly568Val.
CASE REPORT
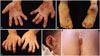
A 3-year-old girl visited our department with severe bilateral palmoplantar hyperkeratosis. Thick, yellowish hyperkeratotic plaques with a foul odor were found on both palms and soles (Fig. 1A and B). She also had thin, brittle nails on all fingers and toes (Fig. 1C), and thin and sparse scalp hairs. The hyperkeratotic lesions initially developed as several punctuate hyperkeratotic plaques, gradually extending to the entire palmoplantar surface. There was yellowish hyperkeratosis on her gluteal fold (Fig. 1D). Multiple hyperkeratotic papules with erythematous, eczematous patches were noted around her eyes, ears and nose (Fig. 1E). She complained of intense pruritus, causing sleep disturbance. She also had walking difficulty due to pain of the plantar lesions.
Her height and weight were below the 3rd percentile for her age. Complete blood counts and routine chemistry panels revealed no abnormalities. Her parents were unaffected. A skin biopsy from the coccygeal lesion revealed hyperkeratosis, occasional parakeratosis, hypergranulosis, acanthosis, papillomatosis and follicular plugging with dilated vessels and inflammatory infiltrates of lymphocytes, histiocytes and eosinophils (Fig. 2A and B) in the upper dermis. Toluidine blue stains showed increased numbers of mast cells in the dermis (Fig. 2C). Electron microscopy showed multiple, variable-sized electron-lucent lipid vacuoles in the hyperkeratotic horny layer (Fig. 2D and E), and numerous polysome-like structures in the upper spinous layer (Fig. 2F).
Genomic DNA was extracted from the peripheral blood of the patient and her parents. Sanger sequencing using primers that spanned all exons and exon-intron boundaries (NM_001258205.1) identified a heterozygous G>T transversion at position c.1703 in exon 13 of TRPV3, which leads to the conversion of a glycine residue to valine (p.Gly568Val) (Fig. 3) (primers are listed in Supplementary Table 1, only online). This mutation was not identified in the unaffected parents, indicating a de novo mutation. We did not discover this mutation on 50 healthy Korean controls. The patient showed moderate improvement after treatment with a systemic acitretin, antihistamine, topical antibacterial, steroid ointment and moisturizer.
DISCUSSION
To our knowledge, this report describes the first Korean case of OS with a rare pathogenic mutation, p.Gly568Val, in the TRPV3 gene. Mutations in TRPV3 causing OS have been identified in five different amino acid residues, comprising p.Gly568, p.Gly573, p.Leu673, p.Trp692, and p.Asn415_Arg416.3456789 TRPV3 contains six transmembrane domains (S1–S6), and p.Gly573 which is within the linker region between S4–S5 is the most commonly reported mutation to date in TRPV3.3 Mutations at codon 568, like our case, have been reported in only four OS cases so far; the heterozygous missense mutations at p.Gly568Val and p.Gly568Asp and a splice site mutation at p.Gly568Cys.91011 The amino acid Gly568 also resides within the linker between S4–S5, but near the boundary of S4 and is highly conserved across several species.10 Previous in silico analysis confirmed that the same amino acid substitution in TRPV3 identified in our patient renders the selectivity filter of this ion channel more hyperpermeable.11 Therefore, this variant can be classified as pathogenic (PS1).12
No consistent genotype-phenotype correlations for mutations in the TRPV3 gene are known yet. Notably, a substantial phenotypic diversity in TRPV3-related OS has been reported, even in families whose members share the same mutation, in terms of the severity and extent of the palmoplantar keratoderma and the presence or absence of other hyperkeratotic lesions.10 Indeed, a brazillian OS patient with p.Gly568Val showed mild phenotype such as focal and mild keratodermas, whereas a Japanese case and our patient harboring the same mutation presented diffuse and severe symptoms.1011 These findings suggest a possibility that additional genetic modifiers or environmental factors may affect the phenotype.
In addition to its role as a thermosensor, TRPV3 plays a significant role in mediating itch and pain sensation and regulating keratinocyte proliferation and differentiation, hair growth, wound healing, and inflammation.13 Several ‘gain-of-function’ mutations in the TRPV3 gene in OS patients were confirmed to exaggerate calcium influx, epidermal turnover, and apoptosis in keratinocytes. Many polysome-like structures that are revealed by electron microscopy may reflect a stimulation of keratinocytes proliferation, which results in the massive acanthosis and hyperkeratosis. In addition, numerous lipid droplets in the corneocytes that are similar to the structural abnormalities in the stratum corneum (SC) of genodermatoses such as Netherton syndrome, lamellar ichthyosis, congenital ichthyosiform erythroderma, and other erythrodermic disorders such as erythrodermic psoriasis may suggest the disturbed cornification with severe perturbation of SC barrier function in OS.1415 Similar to the TRPV3 transgenic mice, the lesional skin of our patient showed increased mast cells which may trigger cutaneous inflammation and severe itch.
Currently, there are no effective treatments for OS. Topical treatments including emollients, keratolytics, retinoids, corticosteroids, systemic retinoids, and methotrexate provide only temporary symptom relief.3 Our case was moderately improved by low-dose systemic retinoids and topical corticosteroid. Recently, fatty acid-derived pro-resolvents have been found to modulate the activities of transient receptor potential ion channels. Bang, et al.16 reported 17(R)-resolvin D1, a member of docosahexaenoic acid-derived series of resolvins, can specifically suppress TRPV3-mediated activity. Specific inhibitors of TRPV3, such as 17(R)-resolvin D1, could be a promising, biocompatible therapeutic approach for OS in the future. Although TRPV3 mutations were identified as a cause of OS, the exact pathomechanism is still poorly understood. Further studies on the pathogenesis of this unique disease might lead to the development of more effective treatments.


 XML Download
XML Download