

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Osteosarcoma (OS), a primary bone malignant tumor, is the second leading cause of cancer-related death in children and young adults.1 Although advancements been made in the diagnosis and treatment of OS, survival rates for metastatic or recurrent OS patients are still very poor.2 Therefore, it is essential and urgent to further explore the mechanisms underlying OS development in order to find out novel diagnostic or prognostic biomarkers and effective therapeutic agents.
A growing amount of evidence indicates that aberrant expression of long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) is closely correlated with the development of various diseases, including OS.34567 Some studies have also suggested that lncRNAs could act as competing endogenous RNAs (ceRNAs) to modulate the expression of miRNAs and miRNAs target genes.89 These lncRNAs were found to exert their functions by miRNA response elements, which could absorb endogenous miRNAs like sponges, thereby relieving the repression effect of miRNAs on their target messenger RNAs (mRNAs).8
Taurine upregulated gene 1 (TUG1), a lncRNA, could act as an oncogene or a tumor suppressor in the development and progression of various cancers. For example, TUG1 has been found to play carcinogenic roles, accompanied by a high-level expression, in some cancers, including esophageal squamous cell cancer and bladder urothelial cancer.1011 However, in some cancers, such as non-small cell lung cancer, TUG1 has been shown to act as a tumor suppressor with low-level expression.12 These studies indicate that TUG1 may be cancer type specific and that different tumor microenvironments might impact TUG1 activity.
In recent years, studies have revealed the critical roles of TUG1 in the progression of OS: Ma, et al.13 reported that TUG1 expression was up-regulated in OS and that high-level expression of TUG1 was closely correlated with poor prognosis and disease status in OS. Moreover, Zhang, et al.14 demonstrated that down-regulation of TUG1 inhibited proliferation and induced apoptosis of OS cells, indicating that TUG1 acts as an oncogene in OS. However, the exact roles and molecular mechanisms of TUG1 underlying OS progression have not been thoroughly elucidated.
In the present study, we identified that TUG1 is highly expressed in human OS tumor tissues, cell lines, and primary OS cells. Moreover, TUG1 facilitated cell proliferation and suppressed apoptosis by sequestering miR-132-3p from its target gene sex determining region Y-box 4 (SOX4) in OS cell lines and primary OS cells.
MATERIALS AND METHODS
Patient tissue samples and OS cell culture
OS tumor tissue and matched adjacent normal tissue were collected from 22 patients diagnosed with primary OS at the First Affiliated Hospital of the Medical College, Shihezi University. This study was performed with the approval of the Research Medical Ethics Committee of the First Affiliated Hospital of the Medical College, Shihezi University. Each patient signed written informed consent prior to enrolling in this medical study.
Human OS cell lines (U2OS, MG-63, Saos-2, and 143B) and the human normal osteoblastic cell line FOB1.19, together with Human Embryonic Kidney 293 cells (HEK293), were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA). U2OS and 143B cells were cultured in RPMI-1640 (Gibco Co., New York, NY, USA) medium supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA). MG-63 were grown in MEM medium (Gibco) containing 10% FBS (Invitrogen). Saos-2 cells were cultured in McCoy's 5A medium (Sigma-Aldrich, St. Louis, MO, USA) containing 15% FBS (Invitrogen). The human normal osteoblastic cell line hFOB 1.19 was maintained in DMEM/F-12 medium (Gibco) supplemented with 10% FBS (Invitrogen). HEK293 cells were maintained in DMEM (Gibco) medium containing 10% FBS (Invitrogen). All cells were maintained in humidified incubator containing 5% CO2 at 37℃.
Establishment of a primary OS cell line
Fresh OS tumor tissue obtained from patients with primary spontaneous OS was washed using sterile phosphate-buffered saline three times and then minced into small tumor pieces. Then, tumor samples were digested in digestion medium [DMEM/F-12 medium (Gibco) containing 0.1 mg/mL of hyaluronidase (Sigma-Aldrich), 2 mg/mL of collagenase A (Sigma-Aldrich), 60 U/mL of nystatin (Sigma-Aldrich), and 0.1 mg/mL of gentamicin (Sigma-Aldrich)] for almost 50 min at 37℃. Then, cells were cultured in DMEM/F12 medium (Gibco) supplemented with 10% FBS (Invitrogen) in a humidified incubator containing 5% CO2 at 37℃.
Reagents and antibodies
Small interference RNA (siRNA) targeting TUG1 (si-TUG1) and its scramble control (si-NC), miR-132-3p mimic and its negative control (miR-NC), miR-132-3p inhibitor (anti-miR-132-3p) and its control anti-miR-NC, and SOX4 siRNA (si-SOX4) and its scramble control (si-NC) were obtained from GenePharma Co., Ltd. (Shanghai, China). TUG1 and SOX4 cDNA sequences were cloned into pcDNA3.1 (Invitrogen) to produce TUG1 and SOX4 overexpression vectors (pcDNA3.1-TUG1 and pcDNA3.1-SOX4), named TUG1 and SOX4.
Cell transfection
The MG-63, U2OS, and primary OS cells were transfected with miRNA, siRNA, miRNA inhibitors, and plasmids by Lipofe-ctamine 2000 (Invitrogen) according to the manufacturer's instructions, and the cells were harvested at the indicated time points after transfection.
RNA extraction and real time PCR analysis
Total RNA from the tissues and cells was obtained by TRIzol solution (Invitrogen) in accordance with the manufacturer's protocols. cDNA was synthesized by the PrimeScript RT reagent kit (TaKaRa, Tokyo, Japan). Quantitative PCR analysis of TUG1 was performed using the SYBR® Premix Ex Taq™ reagent (TaKaRa), with β-actin as an endogenous control. The reverse transcription and quantification of miR-132-3p was carried out using the miDETECTA Track™ miRNA qRT-PCR Starter kit (RiboBio, Guangzhou, China), referring to the manufacturer's protocols, with U6 snRNA as an internal normalization reference.
The real-time quantitative PCR (RT-qPCR) primers sequences were as follows: TUG1, 5′-CTGAAGAAAGGCAACATC-3′ (forward) and 5′-GTAGGCTACTACAGGATTTG-3′ (reverse); β-actin, 5′-AGTGTGACGTGGACATCCGCAAAG-3′ (forward) and 5′-ATCCACATCTGCTGGAAGGTGGAC-3′ (reverse). Reverse transcription primers, 5′-GTCGTATCCAGTGCAG GGTCCGAGGTATTCGCACTGGATACGACCGACCATG-3′ (miR-132-3p); 5′-AAAATATGGAACGCTTCACGAATTTG-3′ (U6). Quantification primers, miR-132-3p, 5′-GCGCGCGTAACAGTCTACAGC-3′ (forward) and 5′-GTCGTATCCAGTGCAGGGTCC-3′ (reverse); U6, 5′-CTCGCTTCGGCAGCACATATACT-3′ (forward) and 5′-CGCTTCACGAATTTGCGTGT-3′ (reverse).
Western blot assays
Cells from different treatment conditions were obtained 48 hours after transfection, and western blot assays were carried out with the SOX4 and β-actin antibodies (Santa Cruz Biotechnology, Dallas, TX, USA) according to manufacturer's protocols. Briefly, MG-63 and U2OS cells were lysed using RIPA lysis buffer (Beyotime, Shanghai, China) containing cocktail (Roche, Mannheim, Germany) to obtain the whole protein. The protein concentration was measured using Pierce™ BCA Protein Assay Kit (Thermo scientific, Rockford, IL, USA). Then, equal weights (50 µg) of proteins were separated using SDS-PAGE gel, followed by being transferred to PVDF membranes (Millipore, Billerica, MA, USA). Next, the membranes were blocked for 1 h with 5% skimmed milk and incubated with appropriate concentrations of antibodies against SOX4 and β-actin antibodies (Santa Cruz Biotechnology) overnight at 4℃. On the next day, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for another 1 h. Finally, protein signals were detected using the BeyoECL Plus kit (Beyotime) and quantified with Image J software (National Institutes of Health, Bethesda, MD, USA).
XTT assays
MG-63, U2OS, and primary OS cells were seeded into 96-well plates and transfected using Lipofectamine 2000 reagent. Then, cell proliferation was assessed using the XTT Cell Proliferation Assay Kit (Abnova, Taipei, Taiwan) following the manufacturer's protocols. Briefly, 10 µL of XTT was added into 96-well plates at indicated time points (0, 24, 48, and 72 h) after transfection and then incubated for another 3 h at 37℃ incubator. Finally, absorbance was measured at the wavelength of 450 nm.
Luciferase assays
Partial DNA sequences of TUG1 and SOX4 3'-UTR containing wild-type (WT) or mutant (MUT) miR-132-3p binding sites were amplified by PCR and then cloned into pmiR-RB-REPORT™ vectors (RiboBio) to produce WT-TUG1, MUT-TUG1, WT-SOX4-3'UTR, and MUT-SOX4-3'UTR reporter plasmids. Then, constructed reporter plasmids were respectively co-transfected with miR-132-3p or miR-NC into 293T cells using Lipofectamine 2000 (Invitrogen). Then, the luciferase activity in cells from different treatment conditions was measured by the Dual-Luciferase® Reporter Assay kit (Promega, Madison, WI, USA) according to manufacturer's instructions.
Apoptosis rate analysis
After transfection for 48 h, transfected cells were treated using the AnnexinV-FITC/PI Apoptosis Detection Kit (BD Biosciences, Franklin Lakes, NJ, USA), referring to manufacturer's instructions, and then, flow cytometry (FACScan; BD Biosciences) was used to determine cell apoptosis rates.
RESULTS
TUG1 expression is upregulated in OS tumors and cells
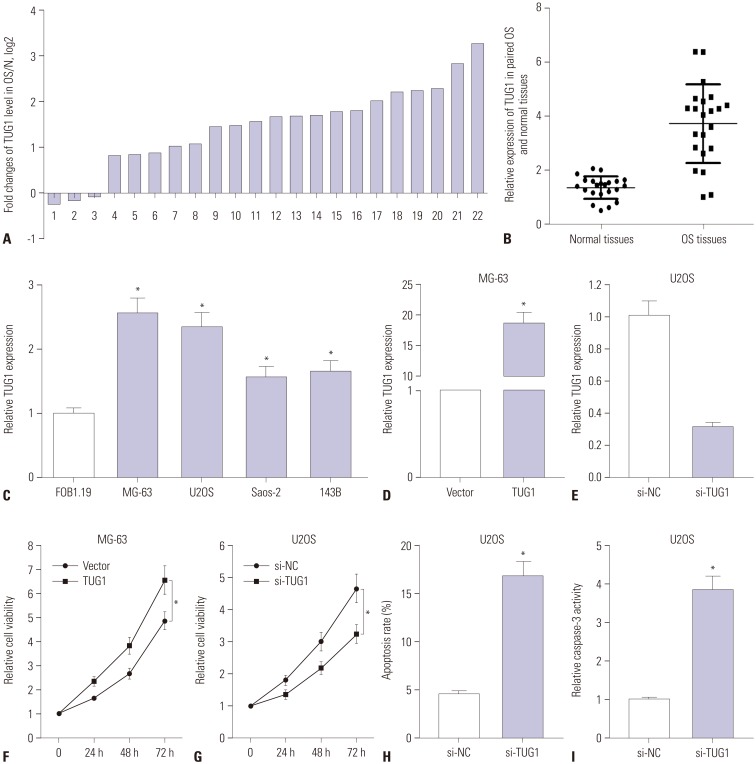
To determine the expression pattern of TUG1 in OS tissues, 22 pairs of OS tumor tissues and correspondingly adjacent normal tissues were collected, followed by the detection of TUG1 levels using RT-qPCR assays. TUG1 expression was remarkably upregulated in 17 (77.3%) OS tumor tissues, compared to correspondingly normal tissues, together with a slight downregulation in the remaining (22.7%) OS tumor tissues (Fig. 1A). The overall expression of TUG1 was upregulated in OS tumor tissues, compared with normal tissues (Fig. 1B). RT-qPCR results revealed that TUG1 was highly expressed in OS cell lines (MG-63, U2OS, Saos-2, and 143B), compared with the human osteoblast cell line hFOB 1.19 (Fig. 1C). All these results indicated that the expression of TUG1 in OS tumors and cells is significantly upregulated, which is in accordance with previous results.1415
TUG1 knockdown suppresses proliferation and induces apoptosis in OS cells (MG-63 and U2OS)
To investigate the roles of TUG1 in OS cell lines, TUG1 overexpression plasmid (pcDNA3.1-TUG1, TUG1) and siRNA of TUG1 (si-TUG1) were constructed or synthesized. Then, the transfection efficiency of TUG1 and si-TUG1 was respectively assessed by RT-qPCR assays. As shown in Fig. 1D, the transfection of pcDNA3.1-TUG1 resulted in a noticeable upregulation of TUG1 expression, compared with the negative control (vector), in MG-63 cells. We also observed that TUG1 expression was strikingly downregulated in U2OS cells transfected with si-TUG1 in comparison with the transfection of non-specific siRNA (si-NC) (Fig. 1E). These results revealed that the TUG1 overexpression plasmid and TUG1 siRNA could exert its function in the following experiments.
Next, the effects of TUG1 on the proliferation and apoptosis of MG-63 and U2OS cells were further investigated. TUG1 and si-TUG1 were respectively transfected into MG-63 and U2OS cells, and cells proliferation was assessed at 0, 24, 48, and 72 h by XTT assays (Fig. 1F and G). The results revealed that TUG1 overexpression significantly promoted MG-63 cell proliferation (Fig. 1F) and that TUG1 knockdown markedly suppressed U2OS cell proliferation (Fig. 1G). The effect of TUG1 on U2OS cell apoptosis was confirmed by the flow cytometry and caspase-3 activity detection. As expected, the depletion of TUG1 by si-TUG1 induced increases in apoptosis (Fig. 1H) and caspase-3 activity (Fig. 1I) in U2OS cells.
Si-TUG1 exerts its anti-proliferation and pro-apoptosis effects partly by targeting miR-132-3p in OS cells
Emerging evidence indicates that lncRNAs act as ceRNAs of miRNAs to regulate target mRNAs expression.89 Therefore, bioinformatics prediction analysis was performed by the miRcode online website to search for potential target miRNAs of TUG1. Among candidate miRNAs, miR-132-3p was chosen due to its anti-tumor effect in OS (Fig. 2A).1617 For validation thereof, WT-TUG1 (containing the putative miR-132-3p binding sites) and MUT-TUG1 (putative miR-132-3p binding sites mutated to the indicated sequences) luciferase reporter plasmids were constructed. Then, the effect of miR-132-3p on the luciferase activity of the WT-TUG1 or MUT-TUG1 reporter plasmid was detected, and the results indicated that the introduction of miR-132-3p mimic significantly inhibits the luciferase activity of WT-TUG1 reporter, compared with miR-NC, whereas miR-132-3p had no effect on the luciferase activity of MUT-TUG1 reporter in HEK293 cells (Fig. 2B). These finding suggested that the putative binding sites were indeed essential for the direct interaction of miR-132-3p and TUG1. RT-qPCR assays further demonstrated that enforced expression of TUG1 markedly suppressed miR-132-3p expression in MG-63 cells, while TUG1 knockdown facilitated miR-132-3p expression in U2OS cells (Fig. 2C). Thus, TUG1 inhibits the expression of miR-132-3p by direct interaction in MG-63 and U2OS cells.
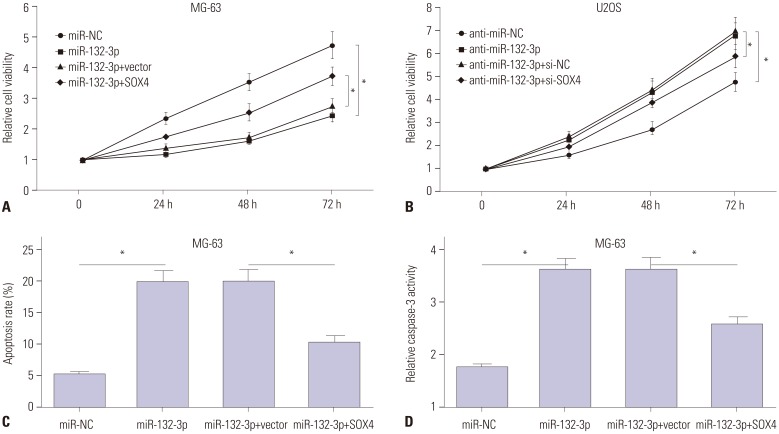
Next, we further explored whether miR-132-3p could influence the effect of TUG1 on proliferation and apoptosis in OS cells. XTT assay results indicated that the restoration of miR-132-3p partly abolished the promotion effect of TUG1 overexpression on proliferation in MG-63 cells (Fig. 2D). Also, the introduction of miR-132-3p inhibitor partially abrogated si-TUG1-mediated anti-proliferative effects in U2OS cells (Fig. 2E). Moreover, the transfection of miR-132-3p inhibitor resulted in a marked decrease in apoptosis rate (Fig. 2F) and caspase-3 activity (Fig. 2G) in U2OS cells upon transfection of si-TUG1, compared to U2OS cells transfected with si-TUG1 and control (anti-miR-NC). Collectively, si-TUG1 exerted its anti-proliferation and pro-apoptosis effects partly by targeting miR-132-3p in OS cells.
SOX4 is a target of miR-132-3p
As is known, miRNAs can exert regulatory roles by affecting target gene expression. Hence, TargetScan online was employed to search for genes that had a chance to interact with miR-132-3p. Among candidate genes, SOX4 has been identified as an oncogene in OS (Fig. 3A),161819 and was chosen for further study. To validate whether the putative binding sites of SOX4-3'UTR and miR-132-3p are important for the interaction between miR-132-3p and SOX4, dual luciferase reporter assays and mutation assays were performed. Then, constructed WT-SOX4-3'UTR or MUT-SOX4-3'UTR reporter was co-transfected with miR-132-3p or miR-NC into HEK293 cells, and their luciferase activities were measured 48 h after transfection. As shown in Fig. 3B, the introduction of miR-132-3p mimic brought about a notable reduction in luciferase activity of the WT-SOX4-3'UTR reporter, compared with negative control (miR-NC). However, miR-132-3p had no effect on the luciferase activity of MUT-SOX4-3'UTR reporter (Fig. 3B). These findings indicated that miR-132-3p interacts with SOX4-3'UTR via the putative binding sites. Next, western blot assays further suggested that the ectopic expression of miR-132-3p decreases SOX4 protein levels, compared with the negative control miR-NC, in MG-63 cells (Fig. 3C). Conversely, miR-132-3p deficiency resulted in a marked increase in SOX4 protein expression, compared with control (anti-miR-NC), in U2OS cells (Fig. 3C). Overall, these results suggest that SOX4 is a target of miR-132-3p in OS cells.
MiR-132-3p inhibits proliferation and induces apoptosis by targeting SOX4 in OS cells
Then, the effects of miR-132-3p and SOX4 on proliferation and apoptosis were further investigated in MG-63 and U2OS cells. XTT assays showed that the introduction of miR-132-3p mimic remarkably suppressed MG-63 cell proliferation, compared with negative control (miR-NC) (Fig. 4A). As expected, the transfection of miR-132-3p inhibitor increased U2OS cell proliferation, compared with negative control (anti-miR-NC) (Fig. 4B). Moreover, the overexpression of miR-132-3p gave rise to a notable increase in apoptosis rate (Fig. 4C) and caspase-3 activity (Fig. 4D) in MG-63 cells. In restoration assays, the reintroduction of SOX4 partly reversed miR-132-3p-mediated anti-proliferation (Fig. 4A) and pro-apoptosis (Fig. 4C and D) effects in MG-63 cells. Also, the introduction of si-SOX4 weakened the promotive effect of miR-132-3p inhibitor on proliferation in U2OS cells (Fig. 4B). Collectively, miR-132-3p was deemed to inhibit proliferation and induce apoptosis by targeting SOX4 in OS cells.
TUG1 enhances SOX4 expression by acting as a sponge of miR-132-3p
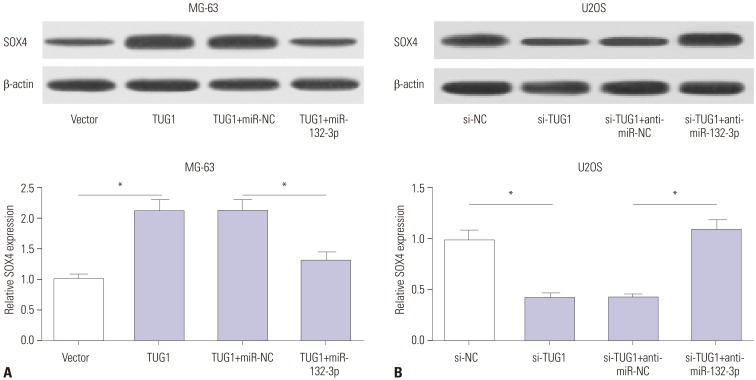
Interestingly, TUG1 and SOX4 shared the same binding sites in the process of interacting with miR-132-3p, indicating that TUG1 might act as a ceRNA to regulate the expression of miR-132-3p and SOX4. Western blot assays demonstrated that TUG1 overexpression markedly improved SOX4 protein levels, while this effect of TUG1 on SOX4 expression was dramatically abated by miR-132-3p in MG-63 cells (Fig. 5A). On the contrary, TUG1 knockdown resulted in a significant reduction in SOX4 protein expression and miR-132-3p inhibitor reversed the inhibition effect of si-TUG1 on SOX4 expression in U2OS cells (Fig. 5B). Taken together, TUG1 might serve as a ceRNA of miR-132-3p to increase SOX4 expression in OS cells.
The knockdown of TUG1 suppresses proliferation and promotes apoptosis by regulating miR-132-3p/SOX4 axis in primary OS cells
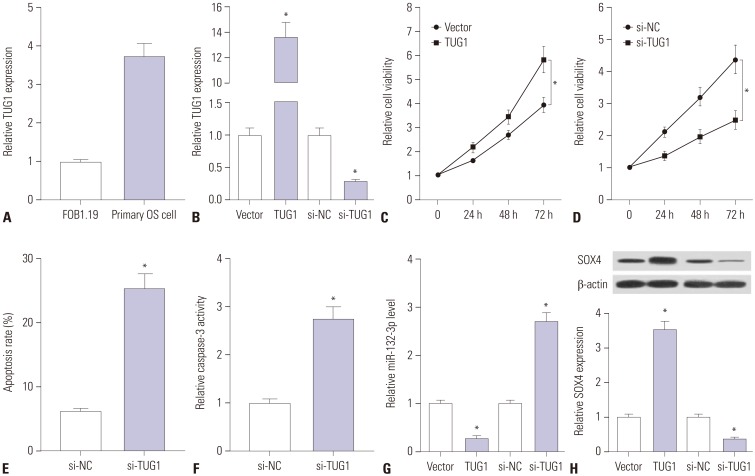
Primary OS cells were isolated from OS patients to further explore whether TUG1 exerts its carcinogenic effect by regulating miR-132-3p/SOX4 in human OS patients. First, we demonstrated that TUG1 is highly expressed in human primary OS cells, compared with the human normal osteoblastic cell line FOB1.19 (Fig. 6A). Next, the transfection efficiency of pcDNA-TUG1 and si-TUG1 was detected in human primary OS cells. As shown in Fig. 6B, the transfection of pcDNA-TUG1 overexpression plasmid strikingly promoted TUG1 expression and the introduction of si-TUG1 markedly inhibited TUG1 expression in human primary OS cells. Then, the effects of TUG1 overexpression and silencing on cell proliferation were further examined in human primary OS cells. The results showed that cell proliferation capacity was elevated in TUG1-overexpressed primary OS cells, which was markedly reduced in TUG1-depleted primary OS cells (Fig. 6C and D). Moreover, we further demonstrated that TUG1 knockdown facilitated primary OS cell apoptosis, reflected as increases in apoptosis rate and caspase-3 activity in si-TUG1-transfected cells (Fig. 6E and F). As demonstrated above, TUG1 exerted its carcinogenic effect by downregulating miR-132-3p expression and upregulating SOX4 expression in OS cell lines. Thus, the effects of TUG1 on miR-132-3p and SOX4 expression were explored in primary OS cells. The results revealed that enforced expression of TUG1 suppresses miR-132-3p expression and facilitates SOX4 expression in primary OS cells (Fig. 6G and H). Conversely, TUG1 silencing induced a notable increase in miR-132-3p level and a marked reduction in SOX4 expression in primary OS cells (Fig. 6G and H). Overall, these data indicated that the knockdown of TUG1 suppresses proliferation and promotes apoptosis by regulating the miR-132-3p/SOX4 axis in primary OS cells.
DISCUSSION
OS, a frequent malignant bone tumor derived from primitive mesenchymal cells, seriously threatens the health of humans, especially madolescents.20 LncRNAs have been identified as critical mediators in the progression and prognosis of OS:721 for instance, lncRNA MALAT1 facilitated metastasis and proliferation of OS cells by the PI3K/AKT axis,22 while lncRNA HIF-2PUT hindered proliferation and invasion of OS cells.23 In the present study, the roles and molecular mechanisms of TUG1 underlying OS development were further investigated. Firstly, RT-qPCR results showed that TUG1 expression is upregulated in OS tumor tissues and cell lines, in accordance with previous reports.1315 Functional analyses further revealed that TUG1 knockdown suppresses proliferation and facilitates apoptosis of MG-63 and U2OS cells, which was also consistent with preceding studies.1415
Herein, bioinformatics predict analysis, luciferase reporter assays, and RT-qPCR assays revealed that TUG1 inhibits miR-132-3p expression by direct interaction. Previous studies have suggested that miR-132-3p might act as a tumor suppressor in OS. For example, Liu, et al.16 demonstrated that miR-132-3p hampered proliferation and metastasis by downregulating target gene SOX4 expression in OS cells. Wang, et al.17 demonstrated that miR-132-3p inhibited OS cell proliferation by targeting cyclin E1. Moreover, Yang, et al.24 reported that low-level expression of miR-132 is associated with poor survival and prognosis in OS patients. Hence, we supposed that TUG1 might exert its carcinogenic effect by downregulating miR-132-3p expression in OS. Indeed, subsequent assays further demonstrated that introduction of miR-132-3p inhibitor partially reversed the effect of TUG1 depletion on proliferation and apoptosis of OS cells. In other words, TUG1 exerted its pro-proliferation and anti-apoptosis effect by targeting miR-132-3p in OS cells.
Mounting evidence suggests that miRNAs exert functions by regulating target mRNAs expression.25 Hence, TargetScan online was employed to search for genes that possibly interact with miR-132-3p. The results indicated that there are some complementary sites between miR-132-3p and SOX4 3'UTR region. SOX4 has been identified as an oncogene in various cancers, including hepatocarcinoma, prostate cancer, and OS.162627 For example, SOX4, as a target of microRNA-212 and microRNA-25-3p together with miR-132, could weaken these miRNAs-mediated inhibition effects on progression of OS cells.161819 Consequently, we further demonstrated that SOX4 was a target gene of miR-132-3p by bioinformatics analysis, luciferase reporter assays, and western blot assays, the results of which were consistent with those in a previous study.16 Additionally, miR-132-3p repressed proliferation and induced apoptosis of OS cells, while this effect of miR-132-3p was partly abrogated by SOX4 overexpression.
Previous studies suggested that TUG1 could act as ceRNA to influence the expression of miRNA target genes. For example, Xie, et al.28 demonstrated that TUG1 promotes the occurrence and progression of OS by acting as an endogenous sponge of miR-9-5p and regulating the expression of the miR-9-5p target gene POU2F1. In addition, Wang, et al.29 also demonstrated that TUG1 promotes the migration and invasion of OS cells by acting as an endogenous sponge of miR-335-5p and by regulating the expression of the miR-335-5p target gene ROCK1. Therefore, we aimed to further explore whether TUG1 could act as a ceRNA to sequester miR-132-3p from its target gene SOX4 in OS. Interestingly, we noticed that TUG1 and SOX4 shared the same binding sites in their interactions with miR-132-3p, indicating that TUG1 might regulate SOX4 expression. Then, we confirmed that TUG1 facilitated SOX4 expression in OS cells. Furthermore, the reintroduction of miR-132-3p partially abrogated TUG1-induced SOX4 up-regulation in OS cells. In other words, our study demonstrated that TUG1 acted as a ceRNA of miR-132-3p to promote SOX4 expression and inhibit miR-132-3p expression, which resulted in an enhancement of proliferation and a reduction of apoptosis in OS cell lines. Additionally, we showed that TUG1 knockdown suppresses proliferation and promotes apoptosis by upregulating miR-132-3p and downregulating SOX4 in human primary OS cells, indicating that TUG1 might exert its carcinogenic effect in OS patients.
Previous studies reported that the promoter of TUG1 encompasses an abundance of highly conserved binding sites of p53 and that its expression is regulated by p53.30 Due to the important roles of p53 in various respects of cancer progression, our study is vitally important and meaningful for investigating therapy and prognosis values of TUG1, miR-132-3p, and SOX4 in multiple cancers, including OS.


 XML Download
XML Download