

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Although it is not common, very high triglyceride (TG) levels (>885 mg/dL) may cause clinical complications, and effective therapeutic approaches for this metabolic disorder are under investigation.1 Although previous studies have described genetic information of patients with very high TG, data from Asian countries are highly limited.234 Particularly, the prevalence and characteristics of patients in Korea have not been analyzed before. The aim of this study was to investigate the prevalence and characteristics of variants of five lipolysis-related genes in Korean patients with very high TG. We selected 26 patients meeting the lipid criteria from 13545 people and performed targeted next generation sequencing of LPL, APOC2, GPIHBP1, APOA5, and LMF1 to identify rare and common variants.
Unrelated patients with very high TG levels were included in this study. Between November 2000 and March 2011, 13545 subjects were enrolled in the Cardiovascular Genome Center Cohort, Yonsei University College of Medicine, Seoul, Korea. Men and women were recruited in the cohort when they visited Severance Hospital, Seoul, Korea for cardiovascular diseases or health check-ups. Patients were interviewed about their medical histories, and then underwent physical examinations. Among them, 26 subjects with fasting TG >885 mg/dL, documented at least two times, were finally included in this study. Patients with obesity (body mass index >30 kg/m2), uncontrolled diabetes mellitus (HbA1c ≥8%), excessive alcohol consumption (>15 drinks/week for men or >8 drinks/week for women), hypothyroidism, proteinuria, pregnancy, or corticosteroids or oral estrogen were excluded. All participants gave their written informed consent and Institutional Review Board of Severance Hospital approved the study (4-2001-0039).
Five target genes were sequenced: LPL (MIM 609708), APOC2 (MIM 608083), GPIHBP1 (MIM 612757), APOA5 (MIM 606368), and LMF1 (MIM 611761). Genomic DNA from blood was extracted with the Qiagen DNeasy kit (Qiagen, Valencia, CA, USA). For mutation analysis, a panel for targeted DNA capture and sequencing by selecting five genes associated with lipolysis was developed by Celemics, Inc. (Seoul, Korea). For targeted sequencing, DNA fragments containing all coding exons and exon-intron junctions were enriched by solution-based hybridization capture, followed by sequencing with an Illumina Hiseq2000 platform (Illumina, San Diego, CA, USA). Analysis of next-generation sequencing data was performed using an in-house analysis pipeline. Variants were called using the GATK v3.3.0 Unified Genotyper algorithm (Broad Institute, Cambridge, MA, USA) for loci with sequencing depth greater than or equal to 20X. Functional annotation of genetic variants was performed by ANNOVAR (ver. 2014-11-12).
Functional effect predictions for single nucleotide variants were performed using SIFT, PolyPhen-2, and MutationTaster, and were matched against Korean population exome data (n=476) and public databases of variants (dbSNP 138, Exome Variant Server and 1000 Genome project SNP from both Asian and all-population databases). Analyses comprise evolutionary conservation, splice-site changes, loss of protein features, and changes that might affect the amount of mRNA. Test results are then evaluated by a naïve Bayes classifier, which predicts disease potential. We then prioritized variants according to the following criteria: 1) variants that were reported to be disease-causing in the Human Gene Mutation Database; 2) disruptive variants (nonsense, splice-site, and frameshift) that were novel or rare (minor allele frequency <1% in public databases); and 3) novel or rare missense variants that were predicted to be deleterious by SIFT, Polyphen-2 (HumVar), or MutationTaster. Variants that met these criteria were validated by bidirectional Sanger sequencing of PCR amplicons.
Clinical characteristics of the study patients are described in Table 1. They were younger than the total population, and more likely to be male (85%). Among 26 subjects, five (19%) had a history of coronary artery disease, whereas one (4%) had a history of pancreatitis. The median TG level was 1213 mg/dL, while the mean high-density lipoprotein-cholesterol was 32.9 mg/dL.
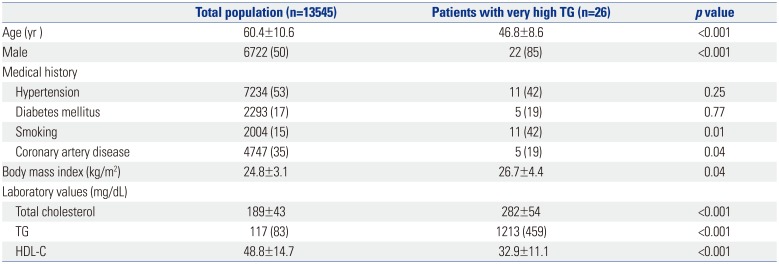
Table 1
Clinical Characteristics of Study Patients
![]()
Ten rare variants of three candidate genes were found in nine (34.6%) patients (three in LPL, four in APOA5, and three in LMF1) (Tables 2 and 3). Six were novel and all variants were suspected of being disease-causing. Nine of them were heterozygous, and one (3.8%) subject had homozygous form of a rare variant in LPL. Six common variants of four genes were observed in 25 (96.2%) patients (one in LPL, one in GPIHBP1, two in APOA5, and two in LMF1) and have been previously reported. Among common variants, c.G41T (p.C14F) of GPIHBP1 and c.G533T (p.G185C) of APOA5 were most frequent, and were found in 15 (57.7%) and 14 (53.8%) patients, respectively. One (3.8%) patient did not show any rare or common variant. In addition, no variant was found in APOC2 (Fig. 1).
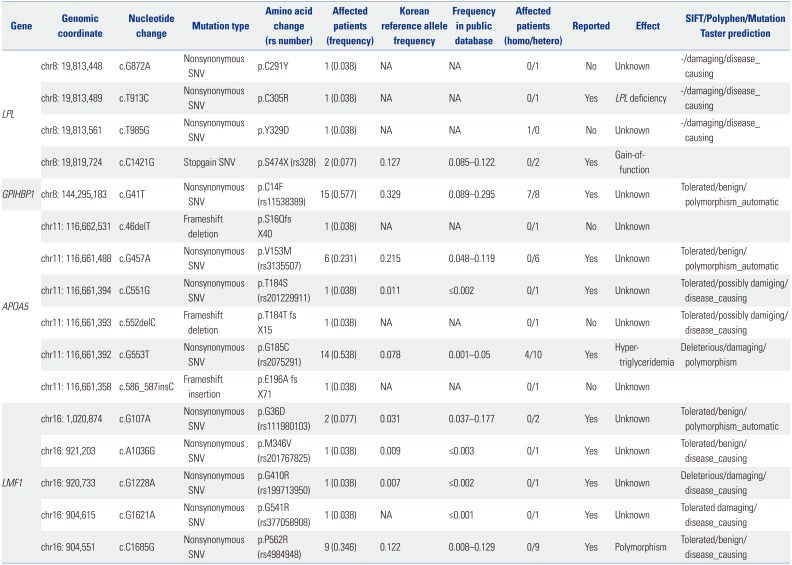
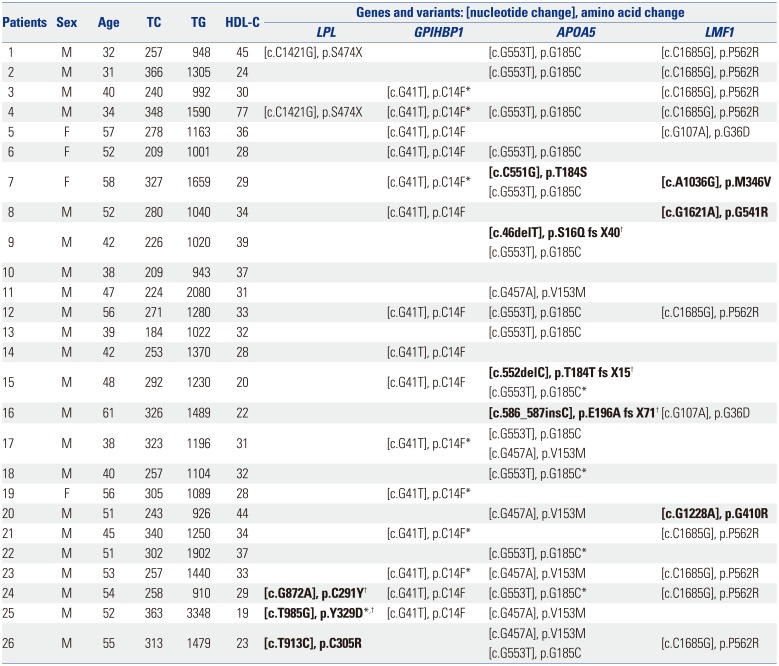
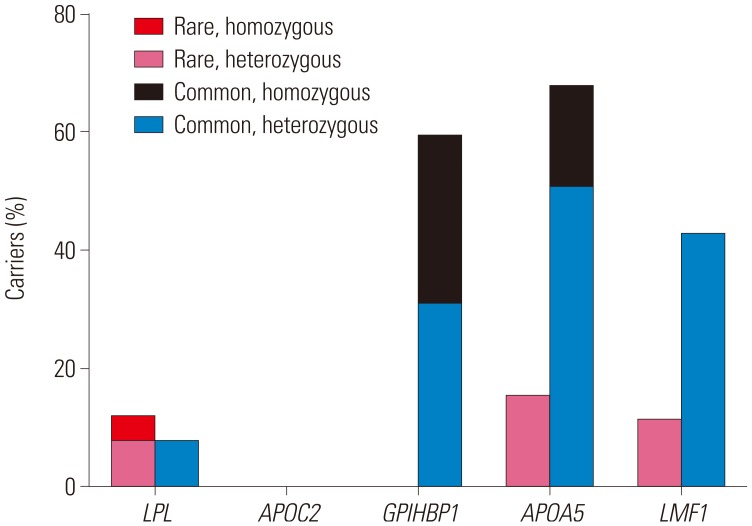
Table 2
Genetic Variants Iidentified in Candidate Genes in Study Patients
![]()
Table 3
Genetic Variants of Target Genes Identified in Each Individual
![]()
Three rare variants of LPL, c.G872A (p.C291Y), c.T913C (p. C305R), and c.T985G (p.Y329D), were identified in three subjects. The c.G872A and c.T985C variants were novel and were predicted to be damaging by in silico analysis, while c.T913C variant has been known to be associated with LPL deficiency.5 Only the c.T985G variant was homozygous. One common variant that has been reported as a stopgain single nucleotide variant,6 c.C1421G (p.S474X), was found in two patients.
One common variant of GPIHBP1, c.G41T (p.C14F), was found in 15 patients and seven of them were homozygous. This variant has been reported previously, but its influence on protein function has been only partially shown.78
In silico algorithms indicated that this variant could be benign.
Four rare variants of APOA5, c.46delT (p.S16Q fs X40), c.C551G (p.T184S), c.C552celC (p.T184T fs X15), and c.586_587insC (p.E196fs), were discovered in four subjects. Patients possessing these variants were all heterozygous, and three of four variants have not been reported before. The c.46delT and c.552delC were frameshift deletions; c.586_587insC was a frameshift insertion; and c.C551G was a single nucleotide variant. These four rare variants were assumed to be disease-causing in prediction algorithms. Two common variants of APOA5, c.G457A (p.V153M) and c.G553T (p.G185C), were found in six and 14 people, respectively. These two variants have been previously reported. c.G553T has been known to be associated with hypertriglyceridemia,39 while c.G457A has been reported to be benign.10
Three rare variants of LMF1, c.A1036G (p.M346V), c.G1228A (p.G410R), and c.G1621A (p.G541R), were identified in three patients. They were all heterozygous and already have been reported. In silico analysis indicated that these three variants may be disease-causing. Two common variants, c.G107A (p. G36D) and c.C1685G (p.P562R), were discovered in two and nine individuals, respectively. Both of these single nucleotide variants were suspected of being benign in prediction algorithms.
The major findings of our study are: 1) 10 rare variants of three genes were found in nine (34.6%) patients (three in LPL, four in APOA5, and three in LMF1). Among them, five were novel and nine were heterozygous. All rare variants were suspected of being disease-causing by in silico analysis. 2) Only one (3.8%) subject had the homozygous form of a rare variant of LPL. 3) Six common variants of four genes were identified in 25 (96.2%) patients (one in LPL, one in GPIHBP1, two in APOA5, and two in LMF1). 4) c.G41T of GPIHBP1 and c.G553T of APOA5 were the most frequent common variants, found in 15 (57.7%) and 14 (53.8%) patients, respectively. We showed, for the first time, the prevalence and characteristics of rare and common variants of five lipolysis-related genes in Koreans with very high TG.
To date, more than a hundred homozygous or compound heterozygous mutations have been identified and shown to cause LPL deficiency. Additional heterozygous mutations have also been found to reduce LPL activity.211 In our results, one rare variant (3.8%), c.T985C, was homozygous, and two rare variants (7.7%), c.G872A and c.T913C, were heterozygous. Analyses by multiple algorithms predicted that these variants may be disease-causing. In patients with severe hypertriglyceridemia of other ethnicities, homozygous or compound heterozygous rare variants of LPL were in 1% in Thai study,4 20% in Dutch study,12 and 23% in Italian study.13 Conversely, heterozygous rare variants were 6% in Canadian and Dutch studies,1214 10% in Italian study,13 and 11% in Thai study.4 The prevalence of such variants in our study was lower than in those studies; nonetheless, homozygous variants appear scarce in Asian studies, including ours. It is worth mentioning that loci of rare variants of LPL are quite diverse. Only one common variant of LPL, c.C1421G, was discovered in two of our subjects. However, it is uncertain whether this variant is associated with hypertriglyceridemia, because these patients had other common functional variants, such as c.G553T. In addition, the relationship between the variant and phenotype was not consistent in prior studies.415
Although rare variants of GPIHBP1 are steadily being discovered,1216 only one form of the common variant was identified in our study. The biological impact of our variant, c.G41T, is only partially known. This variant was identified in 11% of French patients with severe hypertriglyceridemia, in association with a mutation in another site of GPIHBP1. In that study, the c.G41T variant was shown to be associated with mildly reduced protein levels, and was assumed to influence phenotype.7 This variant was also identified in 26% of Spanish patients with the same lipid trait. However, most patients with this variant simultaneously had other variants related to TG levels, thus the impact of this variant was not sufficiently clear.17 This is in accordance with findings in our subjects who had both the c.G41T variant and other rare and common variants of LPL, APOA5, and LMF1.
Our study identified four (15.4%) patients with four rare variants of APOA5 that were all heterozygous. Three of them were novel and all variants were assumed to be disease-causing in prediction algorithms. Several rare variants of APOA5 have been reported in prior studies, and have been diverse in multiple ethnicities. The prevalence was 1.1% in a study of patients with European ancestry and severe hypertriglyceridemia,18 2.3% in a Dutch study,12 and 9.2% in a German study.10 The prevalence of rare APOA5 variants in our population was relatively higher that of other studies. It is also noteworthy that three of four variants in our study were frameshift substitutions or insertions. However, it is difficult to tell how much of an effect these rare variants had on phenotype. A high prevalence (53.8%) of the common c.G553T variant of APOA5 was another characteristic in our subjects. A previous study showed that heterozygous patients of Asian ancestry with this variant have 2.7 times higher TG levels than controls.9 It is interesting that this variant has also been reported in Chinese, Pacific Islander,9 and Taiwanese populations.3
We identified three rare variants of LMF1, and two (7.7%) of them were predicted to be disease-causing. LMF1 has recently been reported to have a role in lipoprotein lipase maturation.19 To date, although only a few rare variants have been discovered and shown to be relevant to TG levels,16 novel variants of LMF1 are being explored. Rare variants were found in 3.4% of Spanish patients with severe hypertriglyceridemia. In Dutch patients, 9.3% revealed these kinds of variants, although only 3.5% were linked to the phenotype.12 Taken together, the prevalence of rare variants of LMF1 in severe hypertriglyceridemia was largely similar between different ethnicities.
Our study has potential limitations. First, several clinical characteristics of the study population selected for re-sequencing were different from those of the total population. For instance, the mean body mass index and the rate of alcohol intake (data not shown) were slightly higher in the study subjects. We cannot completely rule out the fact that these factors might have induced secondary elevation of TG. However, we tried to minimize these effects by excluding patients with high body mass index, uncontrolled diabetes mellitus, or heavy alcohol intake. Second, it was difficult to include and examine variants in family members of the probands. Because the pathogenicity of variants is occasionally clarified by this process, we could have obtained more concrete information on the variants if it has been performed.
In conclusion, we comprehensively analyzed and reported the prevalence and characteristics of variants of LPL, APOC2, GPIHBP1, APOA5, and LMF1 in Korean patients with very high TG. Rare homozygous variants were very uncommon. According to our data, most study subjects may be manifesting the combined effects of rare heterozygous variants and common variants of these genes.
 XML Download
XML Download