

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common malignancies across the globe, especially in Asia and southern Africa.1 Through a diagnosis followed by systemic examination, HCC can be classified into early, mid, and late (advanced) stages. Surgical resection, liver transplantation,2 or local ablation have generally been used to treat early- and mid-stage HCC, and the 5-year survival rate could be as high as 60–70%.2 However, owing to the lack of effective treatment options and underlying liver disease, patients diagnosed at an advanced stage or with progression experience a much more dismal prognosis after locoregional therapy.3 Until sorafenib was used as a first-line therapy, there was no systemic therapy to improve survival in patients with late-stage HCC.45
Sorafenib, which has anti-proliferative and anti-angiogenic effects, is an oral multikinase inhibitor.6 It can inhibit several cellular signaling pathways, including those of Raf/mitogen-activated protein kinase (MAPK), extracellular signaling-regulated kinase (ERK), as well as the receptors of vascular endothelial growth factor and platelet-derived growth factor.67 As the standard care for patients with advanced HCC, sorafenib has been proven to have the capacity to inhibit HCC cell proliferation, angiogenesis, and so on.89 However, the promising treatment of HCC with sorafenib has limited benefits to survival and very low rates of tumor response, where some patients with HCC even exhibit no initial response to sorafenib,310 indicating the existence of both primary and acquired resistances of HCC cells to sorafenib.9 The primary resistance of HCC to sorafenib has been shown to be due to genetic heterogeneity.10 While sorafenib inhibits several cellular signaling pathways as a multikinase inhibitor, it simultaneously or sequentially activates the addiction switches and compensatory pathways.9 Although several mechanisms have been proposed, as for the acquired resistance of HCC cells to sorafenib, the exact mechanisms remain unclear.1112
The multifunctional cytokine transforming growth factor-β (TGF-β) orchestrates an intricate signaling network to modulate tumorigenesis and progression by exerting a dynamic effect on cancer cells.13 Early in the carcinogenesis process, TGF-β1 suppresses tumors and arrests cell growth, whereas in later and more advanced tumor stages, TGF-β1 potentiates epithelial-to-mesenchymal transition, angiogenesis, tumor progression, invasion, and metastasis.1415 Previous studies have shown that TGF-β1 was overexpressed in HCC cells, and clinical studies showed higher blood levels of TGF-β1 in patients with HCC than in patients with chronic hepatitis or cirrhosis.16 Furthermore, HCC cells with a higher IC50 in response to sorafenib tended to have higher TGF-β1 mRNA expression,16 and TGF-β2 was overexpressed in some HCC cell lines and patient cohorts and a correlation was found between high TGF-β2 expression and lower survival rates (p<0.01).17 Thus, the TGF-β signaling pathway should be explored as a therapeutic target for patients with advanced HCC.
MAPKs are essential components of intracellular signal transduction, in which p38 MAPK plays an essential role in the regulation of gene expression and in controlling cellular responses to the environment, cell growth, and apoptosis. These features have previously made p38 MAPK a molecular target for drug development in the treatment of many human diseases, most notably in the treatment of a variety tumors.18 The involvement of the p38 MAPK cascade in the apoptotic pathway has been demonstrated in two hepatoma cell lines, HepG2 and Huh7, wherein the activation of p38 MAPK inhibited cell growth and induced apoptosis.19 The apoptosis of human hepatoma cell lines results from increased caspase 3 activity via the activation of p38 MAPK: Bid cleavage and cytochrome c release are modulated by p38 MAPK activation.19 In human HCC patients, the activities of p38 MAPK and MKK6 were significantly lower than in adjacent uninvolved liver tissue, whereas the activity of ERK1/2 was significantly increased in malignant lesions.1920 Treatment with sorafenib has been shown to inhibit p38α kinase activity in vitro by targeting the DFG-out conformational state of p38α.21 The reduction of p38 activity as a result of sorafenib treatment could be one of the causes of resistance, therefore, it should be a potential target to induce more effective cell death in HCC patients.
Adenovirus serotype 5 is the most commonly used viral vector in cancer gene therapies human serotype due to their high efficiency, broad range of host transduction, easy genome manipulation, and non-integration into the host genome.22 In this research, therefore, we designed adenovirus-delivered TGF-β short hairpin RNAs (shRNAs) to knock down TGF-β expression. The reduction in TGF-β expression resulted in an increase of p38 activity, thus activating the cellular death signal. When TGF-β shRNA was combined with sorafenib, HCC cells became highly sensitized to sorafenib, and the efficiency of sorafenib against advanced HCC was significantly increased.
MATERIALS AND METHODS
Cell culture
Hep-3B, Huh7, and SK-Hep-1 cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (FBS). SNU-182, SNU-398, and SNU-449 cells were cultured in RPMI with 10% FBS. Cells were maintained in a 37℃ humidified atmosphere containing 5% CO2.
Reagents
Antibodies to GAPDH and sorafenib were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to p38, phospho-p38, phospho-Akt (Ser473), phospho-ERK (Thr202/Tyr204), phospho-p65 (Ser536), phospho-Src (Tyr416), MKK3, and MKK6 were purchased from Cell Signaling Technology (Beverly, MA, USA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Construction of adenoviral vectors
For the expression of siRNAs targeting human TGF-β1 or TGF-β2, each annealed sequences for shRNA were cloned into a pSP72ΔE3-U6/H1 vector after BamHI/HindIII digestion. These vectors, designated pSP72ΔE3-U6-shTGF-β1 and pSP72ΔE3-U6-shTGF-β2 (E3 shuttle vector), were linearized by XmnI digestion, and co-transformed into E. coli BJ5183 together with the SpeI-digested adenoviral vector (dl324-IX) for homologous recombination. The detailed informations about the construction of TGF-β shRNAs was previously described.23 The recombinant adenoviral vectors with abbreviated names are as follows:
NC: Ad-IX-ΔE1--ΔE3, control virus.
shT1: Ad-IX-ΔE1-ΔE3-U6-shTGFβ1, virus expressing shRNA of human TGFβ1 (shTGFβ1).
shT2: Ad-IX-ΔE1-ΔE3-U6-shTGFβ2, virus expressing shRNA of human TGFβ2 (shTGFβ2).
MTS viability assays
The CellTiter 96® Aqueous Assay kit (Promega, Madison, WI, USA) comprises solutions of a novel tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS] and an electron coupling reagent (phenazine ethosulfate). MTS is bioreduced by cells into a formazan product that is soluble in tissue culture media. The conversion of MTS into aqueous, soluble formazan is carried out by dehydrogenase enzymes found in metabolically active cells. Subsequently, HCC cell lines were treated with varying doses of sorafenib (0, 2.5, 5, 10, 15, 20, and 25 µM) in 96-well plates for 24 h. The absorbance of formazan at 490 nm was measured directly from 96-well plates without additional processing, and the quantity of formazan product measured at 490 nm is directly proportional to the number of living cells in culture.
Western blot analyses
Cells were lysed with 1X Laemmli sample buffer (62.5 mM Tris, pH 6.8, 2% sodium dodecyl sulfate, 10% glycerol, 0.002% bromophenol blue) and protein concentration was determined using the BCA Protein Assay Kit (Thermo Scientific, Fremont, CA, USA). Then, protein samples were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and gels were electro-transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). Immunodetection was performed using anti-phosho-Akt (p-Akt), anti-phosho-Src (p-Src), anti-phosho-p65 (p-p65), anti-phoshoERK (p-ERK), anti-phosho-p38 (p-p38), anti-p38, anti-MKK3, anti-MKK6 and anti-GAPDH primary antibodies, with the chemiluminescent and fluorescent image analysis system (Syngene, Cambridge, UK).
Real-time quantitative polymerase chain reactions
Cells were lysed with Trizol reagent (Life Technologies, Carlsbad, CA, USA) and the total RNA was isolated via chloroform extraction. RNA concentration was determined with a Nanodrop 2000 (Thermo Scientific). The real-time polymerase chain reaction (PCR) was performed using a Power SYBR Green RNA-to-CT 1-Step Kit (Life Technologies). The reaction mixture contained the reverse transcriptase enzyme mix, reverse transcription PCR mix, forward primer, reverse primer, RNA template, and nuclease-free water. Human TGF-β1 cDNA was amplified using the forward primer 5′-CAAGGGCTACCATGCCAACT-3′ and the reverse primer 5′-AGGGCCAGGACCTTGCTG-3′. Human TGF-β2 cDNA was amplified using the forward primer 5′-GCTGCCTACGTCCACTTTACAT-3′ and the reverse primer 5′-ATATAAGCTCAGGACCCTGCTG-3′. Human β-actin was amplified using the forward primer 5′-ACTCTTCCAGCCTTCCTT-3′ and the reverse primer 5′-ATCTCCTTCTGCATCCTGTC-3′.
Enzyme-linked immunosorbent assays
Cells were plated in the wells of six-well plates at a density of 1×105 cells/well. Supernatants were collected after 48 hr. The levels of TGF-β1 and TGF-β2 expression were determined by an enzyme-linked immunosorbent assays (ELISA) according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA).
Clonogenic assays
Cells were plated into six-well plates at a density of 1×105 cells/well. Subsequently, HCC cell lines were treated with of various concentrations (0, 2.5, 5, 10, 15, or 20 µM) sorafenib (24 hr) or treated with 2.5 µM sorafenib (12 hr) followed by a pre-treatment with adenoviral vectors (NC, shT1, shT2) for 24 hr. Cells were trypsinized and plated into six-well plates at densities of 5×103 or 1×104 cells/well. They were then monitored daily by microscopy. When cells exhibited colonies, surviving cells were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet.
Animal studies
Tumors were implanted subcutaneously in the abdomen of BALB/c nude mice via the injection of SNU-449 human liver cancer cells (1×107) in 100 µL of Hank's balanced salt solution (Gibco BRL, Carlsbad, CA, USA). When tumors reached a size range of 70–100 mm3, animals were randomized into 5 groups of 8 animals each per group [phosphate buffered saline (PBS), sorafenib, NC+sorafenib, shT1+sorafenib, shT2+sorafenib]. Animals in adenoviral groups or the control group (PBS) were administered adenoviruses intratumorally [virus; 1×109 PFU (plaque-forming units) per tumor in 50 µL of PBS] on days 1, 3, and 5. Sorafenib (30 mg/kg) was administered once daily by gavage for 10 days. The regression of tumor growth was assessed by measuring the length (L) and width (W) of each tumor. Tumor volume was calculated using the following formula: volume=0.52×L×W2.
RESULTS
Sorafenib treatment induced changes in several signaling pathways in HCC cell lines
To evaluate the impact of sorafenib in vitro, we first identified the IC50 of sorafenib in different HCC cell lines. The MTS assay showed that the IC50 of sorafenib in HCC cell lines ranged from 7.5 µM to 15 µM (Fig. 1A). Subsequently, we examined various key signaling pathway molecules, including p-p38, p-p65, p-Src, and p-ERK by using western blot analyses, in HCC cell lines after treatment with sorafenib IC50 concentrations, and observed that phospho-ERK activity was reduced by sorafenib treatment. The activity of p-p38 was also decreased in many HCC cells, which may inhibit the death-related signaling pathway (Fig. 1B). We then performed a clonogenic assay to confirm the cell viability of HCC cells after dose-dependent treatments with sorafenib, and observed that many HCC cells survived treatment, even in the presence of high doses of sorafenib, indicating low sensitivity of HCC cells to sorafenib and their resistance to this form of treatment (Fig. 1C). As shown in Fig. 1D, p-Akt and p-p65 were increased by incubating cell lines with 2.5 µM sorafenib for 24 h, whereas p-p38 levels were also more clearly reduced during the treatment. The increased activities of p-p65 and p-Akt (Fig. 1D) signify that cell lines were in fact resistant to sorafenib. Likewise, the inhibition of p-p38 was observed under conditions of sorafenib resistance, suggesting that it may play a role in inducing resistance to sorafenib in HCC cell lines.
p38-mediated cell death pathway was inhibited by treatment with sorafenib
To increase the activity of p38, we employed the constitutive form of MKK3/6 (MKK3/6E provided by Addgene; Cambridge, MA, USA), which has the ability to induce p38 phosphorylation without any stimulation. Fig. 2A and D show that HCC cell lines transfected with the MKK3/6E plasmid clearly increased p38 phosphorylation (Fig. 2A and D), and the cell viability assay showed that the activation of p38 induced massive cell death in HCC cell lines (Fig. 2B). In addition, when treated with both the MKK3/6E plasmid and sorafenib, cell viability in HCC cell lines was significantly reduced in comparison to cell lines treated with sorafenib only (Fig. 2B). Clonogenic assays also confirmed that cell colonies were reduced by MKK3/6E transfection in comparison to control plasmid, and that fewer cell colonies formed in groups co-treated with MKK3/6E and sorafenib than in cells treated with sorafenib alone (Fig. 2C). In addition, p-Akt and p-p65 were reduced in response to co-treatment with MKK3/6E and sorafenib (Fig. 2D), suggesting that p38 activity, which was inhibited by sorafenib, effectively reduced the cytotoxicity of sorafenib and increased the survival potential of HCC cell lines (Fig. 2C and D). Thus, we can surmise that the inhibition of p38 activity as a result of sorafenib treatment was the underlying mechanism of sorafenib resistance in HCC cell lines, and that an increase in the activity of p-p38 as a result of sorafenib treatment overrode sorafenib resistance in HCC cell lines.
TGF-β expression was reduced by sorafenib treatment in HCC cell lines
In order to clarify the changes in TGF-β expression as a result of sorafenib treatment, we analyzed changes in the mRNA levels of TGF-β by real-time PCR. Fig. 3A show that, while treatment with low concentrations (2.5 µM) of sorafenib did not significantly alter TGF-β expression (p>0.05), the expression was significantly (p<0.05) reduced at high concentrations (20 µM). In addition, TGF-β expression was increased under conditions of sorafenib resistance (Fig. 3B), suggesting that decrease of TGF-β expression could reduce the resistance of HCC cell lines to sorafenib and more effectively induce cell death. Interestingly, when TGF-β expression was knocked down by shRNA, phosphorylation of p38 was also increased (Fig. 3C).
Sorafenib combined with adenovirus expressing shRNA against TGF-β was more effective for inducing cell death in HCC cell lines
Next, we investigated whether the cell death could be increased by low concentrations (2.5 µM) of sorafenib when combined with shRNA against TGF-β. As shown in Fig. 3D and E, phosphorylation of p38 was increased in response to this combined treatment compared to sorafenib-pretreated NC virus-infected control group, and cell viability was lower than in the sorafenibpretreated NC virus-infected control group. These results suggest that TGF-β down-regulation is capable of increasing the cytotoxicity of sorafenib by overcoming sorafenib-induced p38 inactivation.
Anti-tumor effect of sorafenib combined with an adenovirus expressing shTGF-β in xenograft animal models
As described above, a series of in vitro experiments confirmed that an adenovirus expressing shTGF-β increased the activity of p38, thereby decreasing the resistance of HCC cell lines to sorafenib. Subsequently, in order to further confirm the antitumor effect of this combination therapy, and whether this therapy is able to override the resistance of HCC tumor cells to sorafenib, we designed an in vivo experiment in xenograft animal models.
Fig. 4A shows that treatment with adenovirus expressing shTGF-β1 or shTGF-β2 in combination with sorafenib displayed increased anti-tumor abilities in comparison to sorafenib alone. This suggests that while these treatments did effectively reduce the resistance of HCC tumor cells to sorafenib, these treatments did not result in a complete reduction in resistance. Despite these outcomes, we observed no differences in tumor regression in response to shTGF-β1 and shTGF-β2 (Fig. 4A). The survival rate of the animals in our study indicated that the combination therapy of adenovirus expressing shTGF-β1 or 2 with sorafenib was the most effective (Fig. 4B).
DISCUSSION
In the present study, we have demonstrated that a low concentration of sorafenib (2.5 µM) can inhibit the activity of p-p38, however, no significant cell death appeared in the low-dose sorafenib treatment. After the low-dose treatment, the activities of p-Akt and p-p65 were increased. Thus, we can infer that a decrease of p-p38 is the underlying mechanism responsible for sorafenib resistance in HCC cell lines. In other words, sorafenib-induced multikinase inhibition23 can be retarded or even inhibited by p38 inhibition due to the steric hinderance of sorafenib to p38,24 which makes a potential of increase of survival signals such as p65 or src depending on the cellular context. Therefore, after inhibition of p38 activation by sorafenib and resultant acquisition of resistance, survival signals such as p65 or src can be increased indirectly depending on the cancer cell types.
We used MKK3/6E to verify that when the activity of p-p38 is increased, cell death is induced and sorafenib resistance is overcome. As MKK3/6 is upstream of the p38 signal in the signally pathway, its activity can induce the phosphorylation of p38. It has been shown that the structure of MKK3/6E is such that MKK3/6E is able to activate p38 without an external stimulus.25 Our results show that MKK3/6E can significantly improve the activity of p-p38 and induce cell death in a significantly large number of cells (p<0.05). When treatment with MKK3/6E was combined with sorafenib, p-p38 activity was significantly increased in comparison to the activity observed in cells treated with sorafenib alone, but was lower in comparison to the p-p38 activity of cells treated with MKK3/6E alone. MTS and clonogenic assays confirmed that the combined application was better than treatment with sorafenib alone. It can be seen that in response to the low-dose sorafenib treatment, p-p38 activity was reduced, suggesting the mechanism involved in the resistance of HCC cells to sorafenib.
In this study, we observed that cell viability and the expression of TGF-β were increased, when HCC cells exhibited drug resistance after treatment with a low concentration of sorafenib. We have also demonstrated increased activities of p-Akt and p-p65 under conditions of sorafenib resistance, when TGF-β expression was clearly and positively correlated with sorafenib drug resistance. On the other hand, however, TGF-β expression was significantly decreased and cell viability was noticeably inhibited in response to treatment with a high concentration of sorafenib (20 µM). These results indicate that the reduction in cell viability in response to high concentration of sorafenib (20 µM) was associated with decreased TGF-β expression. As a result of this, therefore, we targeted TGF-β, knocking down TGF-β expression by using shRNA in order to improve the rate of cell death and reduce drug resistance.
In this study, the reduced expression of TGF-β by shRNA not only increased the activity of p-p38, but also reduced both cell viability and drug resistance. Although the clonogenic assay showed that the combined application significantly improved cytotoxicity, the treatment, however, did not completely eliminate the formation of cell colonies from every cancer cells examined (Fig. 3E). In order to further clarify the anti-tumor effect, as well as the ability of this combination treatment to overcome drug resistance, therefore, an animal model was established using subcutaneous abdominal injections of an HCC cell line (SNU-449) into BALB/c nude mice. The result showed incomplete regression of tumor in mice, suggesting that we should consider adding a new target in order to further improve cytotoxicity, and to more effectively overcome sorafenib resistance. In the future, the use of oncolytic adenoviruses expressing combined therapeutic genes should be explored, and also the use of immune-competent mouse models, which would be more similar to clinical conditions.
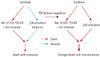
In summary, a series of in vitro experiments demonstrated that the inhibition of p-p38 activity is the underlying mechanism of sorafenib drug resistance, and that TGF-β down-regulation greatly sensitizes the resistance of HCC cells to sorafenib. Fig. 5 provides a schematic diagram representing the results summarized.



 XML Download
XML Download