

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
c-Met, as a tyrosine kinase receptor, is mainly associated with epithelial cell motility, growth and branched tubules formation. Its ligand, the hepatocyte growth factor (HGF), is also known as scatter factor.12 The signaling cascades are triggered through receptor multimerazation, phosphorylation and catalytic activation by combination of HAG and c-Met, and subsequently activates receptor recruiting adaptor proteins such as growth factor receptor-binding protein 2 (Grb2), Src homology and collagen (Shc) and so on, which improves activation of various downstream effector pathways like phosphoinositide-3-kinase (PI3K), mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription (STAT) 3, focal adhesion kinase, and src homology 2-containing tyrosine phosphatase.345
The activation of c-Met stimulates proliferation and anti-apoptotic responses in various types of cells, which mediates epithelial-mesenchymal transition (EMT) and increases motility.67 In cancer, the deregulation of its activity improves cell invasiveness and metastasis by direct involvement of angiogenic pathways.8 According to previous studies, the overex-pression of HGF or c-Met was found in several different types of human cancers, and correlates with tumor metastasis and prognosis of patients.9 Moveover, the c-Met mutations were present in various cancers including papillary renal, ovarian, head/neck, thyroid, liver, and gastric cancers.101112
In non-small cell lung cancer (NSCLC), preclinical evidences have demonstrated the role of c-Met in the pathogenesis, particularly in smoking-driven carcinogenesis.1314
Furthermore, a previous study revealed mutations, overexpression and gene amplification of c-Met in NSCLC patients,15 and the growth of cell lines was suppressed by selective Met small molecule inhibitors or c-Met shRNA.1617 Therefore, c-Met inhibitors could be used as the therapeutic intervention in the treatment of NSCLCs.
The purpose of our study was to identify the inhibited enzymogram and to test the antitumor activity of SIM-89.
MATERIALS AND METHODS
Compounds
SIM-89 (Lot. #1804-06-18, purity 100%) was a kind gift by Simcere Bio-Pharmaceutical Co, Ltd., Staurosporine was purchased from Sigma-Aldrich (USA).
Cell lines and culture
Human NSCLC cell lines H460, and H1299 and adenocarcinoma cell lines A549, H441, and H1993 were purchased from American Type Culture Collection (ATCC, USA). Mouse melanocytoma cell line B16F10 was purchased from KeyGEN Biotech (Nanjing, China). Cells were cultured in RPMI-1640 or DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The FBS, penicillin-streptomycin, cell culture media, and other reagents used in the cell culture were purchased from Invitrogen (Life Technologies, Grand Island, NY, USA). The cell cultures were maintained at 37℃ in a humidified incubator with an atmosphere of 95% air and 5% CO2. HGF was purchased from HumanZyme (Chicago, IL, USA).
Kinase inhibition screening
96-well microplate assays were performed and a positive-control IC50 was used to validate the inhibited kinases. The kinase activities were analyzed by the Invitrogen Z-lyte, LanthaScreen Tb-activity and LanthaScreen Binding assay with 0.01 nmol/L kinase, 200 nmol/L Fluorescein-dual specificity MAPK kinase 1 (MAP2K1), 50 mmol/L Hepes (pH 7.5), 10 mmol/L MgCl2 and 0.01% Brij35 for 1 h at 23℃; the reaction was completed with 30 mmol/L EDTA and 2 nmol/L phosphor-MAP2K1 antibody. For the IC50 test, four serial concentrations of SIM-89 (0.1, 1, 10, and 100 µmol/L) were used, while Staurosporine (10 µmol/L) was used as the positive control. As to MOA study, 6 ATP concentrations (1/3-, 1-, 3-, 9-, 27-, and 81-fold ATP Km) were employed at the same serial dilution used in the IC50 test. PerkinElmer Envision 2104 reader in Htrf/FRET module was used to analyze the readouts. The inhibition % was analyzed using Excel (Microsoft). The IC50 Shift as well as the Lineweaver-Burk double-reciprocal plots were drawn using Graphpad Prism 5. The tested enzymes were summarized in Table 1.
Real time-PCR
Real time-PCR (RT-PCR) assay was used to detect the mRNA levels of three genes, Met, JAK1, and STAT1, in order to ascertain the effect of SIM-89 on cells at transcription level. Thus, A549 (5×103), H1299 (5×103), H441 (3×103), and H460 (1×104) cells were plated in 30-mm dishes for 24, 48, and 72 h, and all cell lines (5×105) were also plated for 2, 4, and 8 h. SIM-89 at 5 µmol/L or 1 µmol/L with or without HGF or vehicle (0.2% DMSO) were used for 2, 4, and 8 h as described above.
Total RNA extraction, cDNA synthesis, and quantitative RT-PCR were performed according to the manufacturer's instructions using RevertAid First Strand cDNA Synthesis Kit (Fermentas). Quantitative analysis of mRNA expression was per-formed using SYBR Green expression assays (Invitrogen, Life Technologies) with ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA).
Expression level of mRNA was calculated utilizing the 2−△△t method with GAPDH as the internal reference. The primers of JAK1, Met, STAT1 and GAPD as listed below: JAK1 (forward: 5′-GAATGACGCCACACTGACTG-3′; reverse: 5′-GATGACAA GATGTCCCTCCG-3′), Met (forward: 5′-TGTTCGATATTCAT CACGGC-3′; reverse: 5′-GCATTTTTACGGACCCAATC-3′), STAT1 (forward: 5′-TGAATATTCCCCGACTGAGC-3′; reverse: 5′-AGGAAGACCCAATCCAGATGT-3′), GAPDH (forward: 5′-CGACCACTTTGTCAAGCTCA-3′; reverse: 5′-AGGGGTCTACATGGCAACTG-3′). All the above primers were purchased from Shanghai Generay Biotech Company (Shanghai, China).
ELISA assay
Appropriate numbers of A549, H460, H1299, H441, and H1993 cells were seeded in 96-well plates and allowed for adhesion overnight. SIM-89 at 5 µmol/L or vehicle (0.2% DMSO) was applied, and the cell culture media were collected at 24, 48, 72, 96, 120, 144, and 168 h, respectively. The levels of HGF in the cell culture medium were measured with ELISA assay (#DHG00, R&D) as the manufacturer's instructions.
Western blotting
The expression and phosphorylation of c-Met were evaluated by western blotting. Cells were washed with ice-cold phosphate buffered saline (PBS) and lysed with lysis buffer (Cell signaling Technology, Danvers, MA, USA) and protease and phosphatase inhibitors as follows: aprotinin (10 mg/mL), leupeptin (10 mg/mL) (ICN Biomedicals, Asse-Relegem, Belgium), phenylmethylsulfonyl fluoride (1.72 mM), NaF (100 mM), NaVO3 (500 mM), and Na4P2O7 (500 mg/mL) (Sigma-Aldrich). The protein concentration was determined by BCA assay (Pierce, USA). Equal amounts of protein were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and subsequently transferred onto polyvinylidene fluoride (PVDF) membrane. Blots immunostaining was performed using the primary antibodies, followed by secondary antibodies conjugated to horseradish peroxidase, and detected by enhanced chemiluminescence reagent (Pierce, USA). The primary antibodies used were anti-phospho-c-Met (pY1230+Y1234+Y1235), anti-phospho-c-Met (pY1349), and c-Met (Epitomics; CA, USA). The secondary antibodies were purchased from Amersham Biosciences (Piscataway, NJ, USA).
CCK8 assay
Cells were seeded on a 96-well plate (Falcon, BD, USA) with 5000–8000 cell density per well, and different concentrations of compounds were added to the culture medium and then incubated for an indicated period. CCK8 assay was used to determine the viability of cell. In brief, 10 µL of CCK8 solution was added to 200 µL of medium in each well, and a microplate reader (Molecular Devices, Hercules, CA, USA) was used to measure absorbance at 450 nm. The inhibition percentage was calculated according to the following formula:

Transwell assay
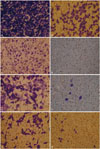
Cells (5×105) were seeded in triplicate in upper chamber of the 12-well tranwell plate (Millipore, 8 µm pore diameter). After adding 500 µL of RPMI-1640 or DMEM+HGF (50 ng/mL)+ series of SIM-89 concentration (50, 10, 2, 0.4, and 0.08 µM) with or without 10% FBS to the lower chamber of the plate, the plate was incubated at 37℃ and 5% CO2 for 24 hrs. Then, the upper chamber was fixed with 95% ethanol for 30 min and stained with crystal violet for 15 min. Subsequently, the membrane was checked with microscopy, and cells in upper chamber were destained with acetic acid and absorbance was read at 570 nm.
RESULTS
Inhibition of receptor tyrosine kinases by SIM-89
The effect of SIM-89 on the enzyme activity was measured after 1 h treatment. The IC50 value of SIM-89 on c-Met, adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) and tyrosine kinase A (TRKA) were 297 nmol/L, 1.31 µmol/L, and 150.2 nmol/L, respectively (Fig. 1, Table 2). The double-reciprocal plot showed linear regression alignments converge on the Y-axis, as shown in Fig. 2A, 2C and 2E. When ATP was added, the IC50 for c-Met increased in consistence with ATP concentration (Fig. 2B), implying that SIM-89 inhibited the activity of c-Met by an ATP-competitive mechanism. For AMPK, the IC50 was in line with the different concentrations (1/3-, 1-, 3-, 9-, 27-, and 81-fold ATP Km) (Fig. 2D), which suggested that SIM-89 suppressed AMPK activity in a non-ATP-competitive mode. As for TRKA, the IC50 increased as the increase of ATP concentration in a non-homogeneous manner (Fig. 2F), which indicated that SIM-89 inhibited the TRKA activity through a mixed-type inhibition. In conclusion, SIM-89 inhibited the activity of kinases, targeting ATP-binding site of kinases, by different mechanisms under cell free conditions.
Inhibitory effect of SIM-89 on cell proliferation
As shown in Fig. 3, a dose dependent inhibition of cell proliferation by SIM-89 was found in A549, H460, H441, H1993, H1299, and B16F10 cells at 24, 48, and 72 h (p<0.05 among all groups with different dose in each cells). The IC50 values for these cell lines at each time points are presented in Table 3: the maximum percent inhibition was 59, 89, 85, 80, 74, and 87% for A549, H460, H441, H1993, H1299, and B16F10 cells, respectively.
Inhibitory effect of SIM-89 on Met, STAT1 and JAK1 expressions in H460 cells
We measured the expression of c-Met in A549, H1299, H441, and H460 cells after the treatment of SIM-89 (Fig. 4A). No significant decrease of c-Met expression was found in A549, H1299, and H441 cells, while significant decrease was found in H460 cells at 8 h after the treatment with 1 µM SIM-89 with (p=0.000) or without (p=0.005) HGF (Fig. 4C). We also examined the expression of STAT1 after the treatment of SIM-89 and found that a significantly decreased expression of STAT1 in 1 µM SIM-89+HGF (p=0.002), 5 µM SIM-89 (p=0.006) and 5 µM SIM-89+HGF (p=0.008) (Fig. 4B). The expression of JAK1 also decreased significantly in 1 µM SIM-89+HGF (p=0.009), 5 µM SIM-89 (p=0.004) and 5 µM SIM-89+HGF (p=0.019) (Fig. 4D). No significant difference was found in the expressions of BRAF, ERK, MAP2K1, and MAP2K2 (Data not shown). These results indicated that SIM-89 suppressed the expressions of Met, STAT1, JAK1 in H460 cells, independent of the Raf-MEK-ERK pathway.
Decreased secretion of HGF in tumor cells after the treatment of SIM-89
We examined the levels of HGF in A549, H441, H460, H1299, and H1993 cells after the treatment of SIM-89. The levels of HGF in A549, H460, H1299, and H1993 cells were decreased after the SIM-89 treatment, suggesting that SIM-89 inhibited the secretion of HGF in tumor cells (Fig. 5).
SIM-89 inhibited c-Met phosphorylation in tumor cells
The phosphorylation of Met was examined in mouse B16F10 cells and human A549, H441, H460, and H1299 cells. We found that SIM-89 decreased the phosphorylation of c-Met (pY1349) and c-Met (pY1230+pY1234+pY1235) in B16F10 cells (Fig. 6A). In A549 (Fig. 6B) and H441 (Fig. 6C) cells, we found a decreased phosphorylation of c-Met (pY1230+pY1234+pY1235). In H460 cells, phosphorylation of both c-Met (pY1349) and c-Met (pY1230+pY1234+pY1235) was increased after SIM-89 treatment (Fig. 6D), whereas the phosphorylation of c-Met (pY1349) was decreased in H1299 cells (Fig. 6E).
The inhibitive effect of SIM-89 on the migration of tumor cells
As shown in Fig. 7, we found a significant decrease of cell migration in A549 (Fig. 7A and B), H441 (Fig. 7C and D), H1993 (Fig. 7E and F), and H1299 (Fig. 7G and H) cells after SIM-89 treatment, and also a significant decrease of EC50 in HGF group compared to FBS group in A549, H1299, and H460 cells (Table 4).
DISCUSSION
Receptor tyrosine kinases, which play an important role in the development and progress of cancer, represent effective targets for specific therapy. Nowadays, receptor kinase-based treatment has widely been used in clinical practice; for examples, in breast cancer (herceptin), in gastrointestinal stromal tumors (gleevec), and in NSCLC (gefitinib).18 However, acquired drug resistance limits the outcome of the therapy. Therefore, novel therapeutic choice is urgently needed.
Over the past decade, there has been considerable data generated on Met biology and therapy for lung cancer. Met receptor tyrosine kinase can be activated through a number of mechanisms, especially with its ligand HGF stimulation. In lung cancer, Met can be overexpressed along with HGF. There can also be gain-of-function mutations within the semaphorin and juxtamembrane domain.192021 In a subset of NSCLC, Met is amplified.222324 In addition, Met and phosphorylated Met (especially juxtamembrane domain site pY1003 and autocatalytic site pY1234/1235) are overexpressed in 40% of tumor tissues, and are prognostic biomarkers in lung cancer.2526 Therefore, Met receptor tyrosine kinase could be a potential target for NSCLC. As an inhibitor targeting Met receptor tyrosine kinase, SIM-89 might be effective in NSCLC. Thus, we performed a series of in vitro studies to explore the anticancer mechanisms of SIM-89.
Ferraro, et al.27 observed that c-Met was overexpressed in a lung-specific (B16F10) metastatic clone of the B16 mouse melanoma and suggested that c-Met exerts a pro-metastatic property in these cells, whereas Navab, et al.28 showed that H460 could spontaneously metastasize to systemic organs through c-Met/HGF related mechanisms. These studies indicate a potential for pro-metastatic property of c-Met and the inhibitory effect of SIM-89 on cell migration.
In the present study, we evaluated the IC50 of SIM-89 by comparison with Staursporing and found a lower IC50 of 297 nmol/L against c-Met. Moreover, SIM-89 showed an inhibitory effect on TRKA and AMPK with IC50 values of 150.2 and 1310 nmol/L, respectively.
Several therapeutic ways could inhibit the activity of protein kinases. Drugs which reversibly bind to ATP-binding site, within kinase domain or to an adjacent small pocket, suppress kinase activity. Due to similarities among three-dimensional structures of the kinase domain, ATP-competitive inhibitors possess cross-reactivity with other kinases of related structures. In the present study, we observed that SIM-89 inhibited the activities of c-Met, AMPK, and TRKA through ATP-competitive, non-ATP-competitive and mixed-type mode, respectively, indicating that SIM-89 inhibited the activity of kinases, targeting ATP-binding site of kinases, by different mechanisms under cell free conditions. Because of it's novelty, further studies are needed to confirm the conclusion.
Since oncogenic mechanisms differ among various cancers, we selected 6 cell lines in the present study, which have different metastatic potentials (A549, H460, H441, H1993, H1299, and B16F10), and found that SIM-89 could suppress the expressions of Met, STAT1, and JAK1 in H460 cells at the transcriptional level, which was independent of the Raf-MEK-ERK pathway. On the other hand, SIM-89 inhibited the phosphorylation of Met (pY1230+pY1234+pY1235) in A549 and H441 cells at the post-transcriptional level. A feedback regulatory mechanism may lead to the increase of expression in the phosphorylation of Met-pY1349. We also found that SIM-89 could directly inhibit the phosphorylation of Met (pY1230+pY1234+pY1235) and Met-pY1349 in B16F10 cells, and that SIM-89 decreased the phosphorylation of Met-pY1349 in the H1299 cells. Therefore, we believe that SIM-89 might exert its antitumor activity by inhibiting the phosphorylation of Met directly at the post-transcriptional level in lung cancer.
In order to explore the signaling pathway affected by SIM-89 treatment, RT-PCR was performed to check the expression level of several intracellular molecules, and found a significant inhibition of the expressions of JAK1 and STAT1 by the treatment of SIM-89. Ravichandran, et al.29 also found the involvement of JAK-STAT in lung cancer growth and progression, consistent with our results. However, we failed to find the involvement of Raf-MEK-ERK pathway. In conclusion, our present study showed that SIM-89 inhibited the proliferation of cancer cells by suppressing related kinases through different competitive mechanisms: c-Met, AMPK and TRKA and the JAK1-STAT1 kinase signaling cascade were crucial targets for anticancer treatment by SIM-89, although with different pharmacological mechanisms involved in different cell lines. However, more studies are needed to further explore detailed mechanisms of SIM-89 involved in the anti-cancer effect, and also to further investigate the association of additional protein kinases with SIM-89 by kinase spectrum screening.









 XML Download
XML Download